Garnica Diana P, Pinzón Andrés M, Quesada-Ocampo Lina M, Bernal Adriana J, Barreto Emiliano, Grünwald Niklaus J, Restrepo Silvia
Laboratorio de Micología y Fitopatología Uniandes (LAMFU), Universidad de Los Andes, Bogotá, Colombia.
BMC Genomics. 2006 Sep 28;7:245. doi: 10.1186/1471-2164-7-245.
Members of the genus Phytophthora are notorious pathogens with world-wide distribution. The most devastating species include P. infestans, P. ramorum and P. sojae. In order to develop molecular methods for routinely characterizing their populations and to gain a better insight into the organization and evolution of their genomes, we used an in silico approach to survey and compare simple sequence repeats (SSRs) in transcript sequences from these three species. We compared the occurrence, relative abundance, relative density and cross-species transferability of the SSRs in these oomycetes.
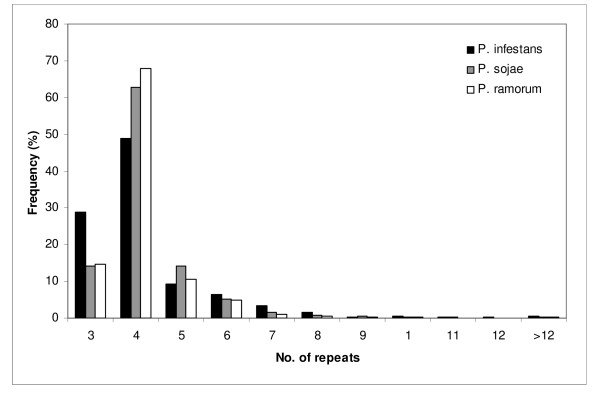
The number of SSRs in oomycetes transcribed sequences is low and long SSRs are rare. The in silico transferability of SSRs among the Phytophthora species was analyzed for all sets generated, and primers were selected on the basis of similarity as possible candidates for transferability to other Phytophthora species. Sequences encoding putative pathogenicity factors from all three Phytophthora species were also surveyed for presence of SSRs. However, no correlation between gene function and SSR abundance was observed. The SSR survey results, and the primer pairs designed for all SSRs from the three species, were deposited in a public database.
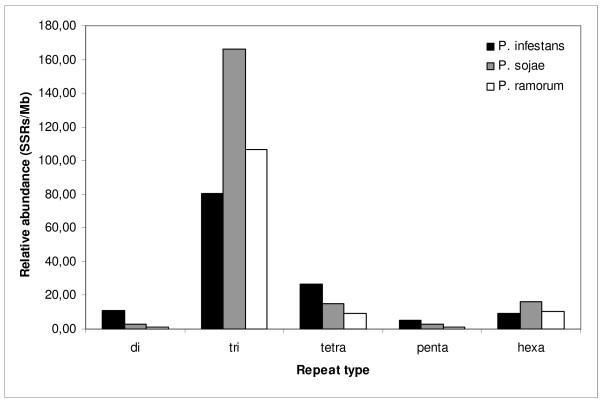
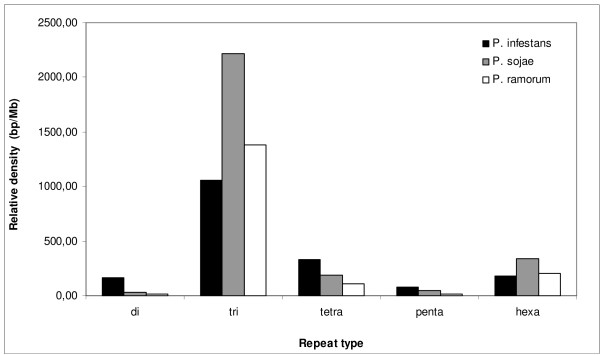
In all cases the most common SSRs were trinucleotide repeat units with low repeat numbers. A proportion (7.5%) of primers could be transferred with 90% similarity between at least two species of Phytophthora. This information represents a valuable source of molecular markers for use in population genetics, genetic mapping and strain fingerprinting studies of oomycetes, and illustrates how genomic databases can be exploited to generate data-mining filters for SSRs before experimental validation.
疫霉属成员是分布于全球的臭名昭著的病原体。最具破坏性的物种包括致病疫霉、栎叶疫霉和大豆疫霉。为了开发用于常规鉴定其种群的分子方法,并更好地了解其基因组的组织和进化,我们采用计算机方法来调查和比较这三个物种转录序列中的简单序列重复(SSR)。我们比较了这些卵菌中SSR的出现情况、相对丰度、相对密度和跨物种转移性。
卵菌转录序列中的SSR数量较少,长SSR很少见。对所有生成的数据集分析了疫霉属物种间SSR的计算机转移性,并基于相似性选择引物,作为可能转移到其他疫霉属物种的候选引物。还调查了所有三种疫霉属物种中推定致病因子编码序列中SSR的存在情况。然而,未观察到基因功能与SSR丰度之间的相关性。SSR调查结果以及为这三个物种的所有SSR设计的引物对已存入公共数据库。
在所有情况下,最常见的SSR是重复次数少的三核苷酸重复单元。一定比例(7.5%)的引物可以在至少两种疫霉属物种之间以90%的相似性转移。这些信息是用于卵菌群体遗传学、遗传图谱绘制和菌株指纹研究的有价值的分子标记来源,并说明了如何在实验验证之前利用基因组数据库生成SSR的数据挖掘筛选器。