Leslin Chesley M, Abyzov Alexej, Ilyin Valentin A
Department of Biology, Northeastern University, 360 Huntington Avenue, Boston, MA 02115, USA.
Nucleic Acids Res. 2007 Jan;35(Database issue):D317-21. doi: 10.1093/nar/gkl809. Epub 2006 Oct 25.
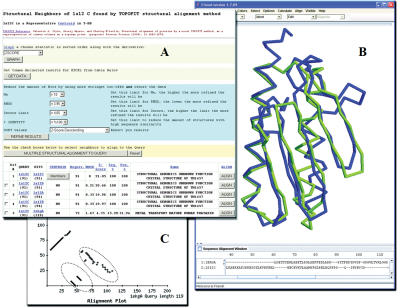
TOPOFIT-DB (T-DB) is a public web-based database of protein structural alignments based on the TOPOFIT method, providing a comprehensive resource for comparative analysis of protein structure families. The TOPOFIT method is based on the discovery of a saturation point on the alignment curve (topomax point) which presents an ability to objectively identify a border between common and variable parts in a protein structural family, providing additional insight into protein comparison and functional annotation. TOPOFIT also effectively detects non-sequential relations between protein structures. T-DB provides users with the convenient ability to retrieve and analyze structural neighbors for a protein; do one-to-all calculation of a user provided structure against the entire current PDB release with T-Server, and pair-wise comparison using the TOPOFIT method through the T-Pair web page. All outputs are reported in various web-based tables and graphics, with automated viewing of the structure-sequence alignments in the Friend software package for complete, detailed analysis. T-DB presents researchers with the opportunity for comprehensive studies of the variability in proteins and is publicly available at http://mozart.bio.neu.edu/topofit/index.php.
TOPOFIT数据库(T-DB)是一个基于网络的公开蛋白质结构比对数据库,它基于TOPOFIT方法,为蛋白质结构家族的比较分析提供了全面的资源。TOPOFIT方法基于在比对曲线上发现一个饱和点(拓扑最大值点),该点能够客观地识别蛋白质结构家族中共同部分和可变部分之间的边界,为蛋白质比较和功能注释提供了更多见解。TOPOFIT还能有效地检测蛋白质结构之间的非顺序关系。T-DB为用户提供了便捷的功能,可用于检索和分析蛋白质的结构邻居;使用T-Server针对当前整个PDB版本对用户提供的结构进行一对多计算,并通过T-Pair网页使用TOPOFIT方法进行成对比较。所有输出结果都以各种基于网络的表格和图形形式呈现,可在Friend软件包中自动查看结构-序列比对结果,以进行完整、详细的分析。T-DB为研究人员提供了全面研究蛋白质变异性的机会,可通过http://mozart.bio.neu.edu/topofit/index.php公开获取。