Abyzov Alexej, Ilyin Valentin A
Department of Biology, Northeastern University 360 Huntington Avenue, Boston, MA 02115, USA.
BMC Struct Biol. 2007 Nov 16;7:78. doi: 10.1186/1472-6807-7-78.
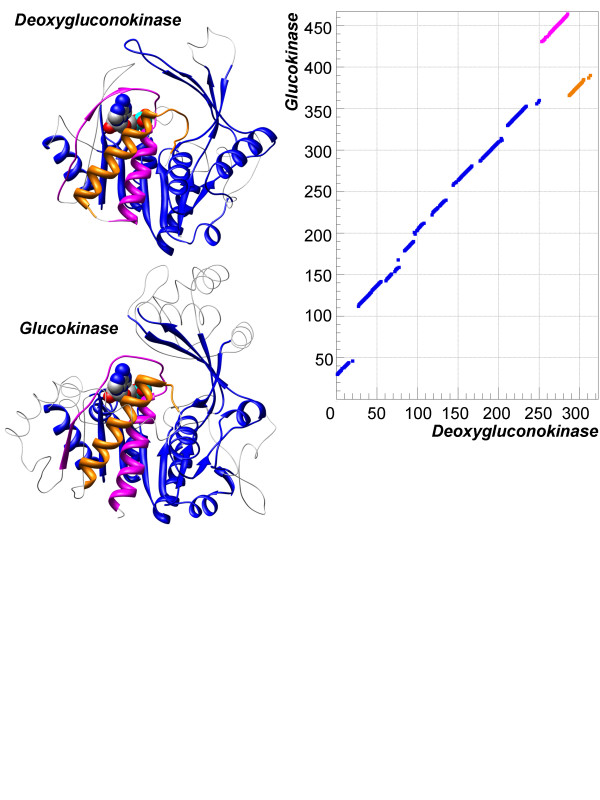
The majority of relations between proteins can be represented as a conventional sequential alignment. Nevertheless, unusual non-sequential alignments with different connectivity of the aligned fragments in compared proteins have been reported by many researchers. It is interesting to understand those non-sequential alignments; are they unique, sporadic cases or they occur frequently; do they belong to a few specific folds or spread among many different folds, as a common feature of protein structure. We present here a comprehensive large-scale study of non-sequential alignments between available protein structures in Protein Data Bank.
The study has been conducted on a non-redundant set of 8,865 protein structures aligned with the aid of the TOPOFIT method. It has been estimated that between 17.4% and 35.2% of all alignments are non-sequential depending on variations in the parameters. Analysis of the data revealed that non-sequential relations between proteins do occur systematically and in large quantities. Various sizes and numbers of non-sequential fragments have been observed with all possible complexities of fragment rearrangements found for alignments consisting of up to 12 fragments. It has been found that non-sequential alignments are not limited to proteins of any particular fold and are present in more than two hundred of them. Moreover, many of them are found between proteins with different fold assignments. It has been shown that protein structure symmetry does not explain non-sequential alignments. Therefore, compelling evidences have been provided that non-sequential alignments between proteins are systematic and widespread across the protein universe.
The phenomenon of the widespread occurrence of non-sequential alignments between proteins might represent a missing rule of protein structure organization. More detailed study of this phenomenon will enhance our understanding of protein stability, folding, and evolution.
大多数蛋白质之间的关系可以用传统的序列比对来表示。然而,许多研究人员报告了一些不寻常的非序列比对,其中比对片段在被比较蛋白质中的连接方式不同。了解这些非序列比对很有意思:它们是独特的、零星的情况,还是经常出现;它们是属于少数特定的折叠类型,还是作为蛋白质结构的一个共同特征分布在许多不同的折叠类型中。我们在此展示了一项对蛋白质数据库中可用蛋白质结构之间非序列比对的全面大规模研究。
该研究基于借助TOPOFIT方法比对的8865个非冗余蛋白质结构集进行。据估计,根据参数的不同,所有比对中17.4%至35.2%是非序列的。数据分析表明,蛋白质之间的非序列关系确实系统且大量地存在。对于由多达12个片段组成的比对,观察到了各种大小和数量的非序列片段,以及片段重排的所有可能复杂性。研究发现,非序列比对并不局限于任何特定折叠类型的蛋白质,而是存在于两百多种此类蛋白质中。此外,其中许多存在于具有不同折叠类型归属的蛋白质之间。研究表明,蛋白质结构对称性并不能解释非序列比对。因此,已经提供了令人信服的证据,证明蛋白质之间的非序列比对是系统的,并且在整个蛋白质世界中广泛存在。
蛋白质之间广泛存在非序列比对的现象可能代表了蛋白质结构组织中一条缺失的规则。对这一现象进行更详细的研究将增进我们对蛋白质稳定性、折叠和进化的理解。