Andreeva Antonina, Prlić Andreas, Hubbard Tim J P, Murzin Alexey G
MRC Centre for Protein Engineering, Hills Road, Cambridge CB2 2QH, UK.
Nucleic Acids Res. 2007 Jan;35(Database issue):D253-9. doi: 10.1093/nar/gkl746. Epub 2006 Oct 26.
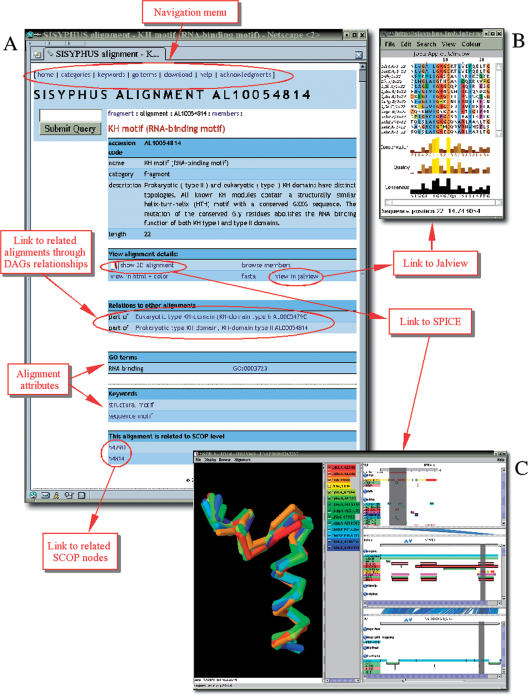
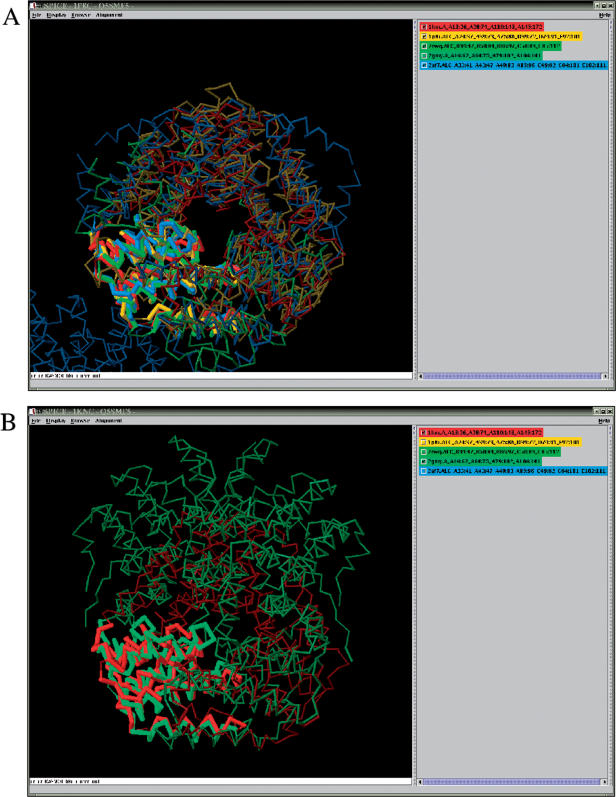
With the increasing amount of structural data, the number of homologous protein structures bearing topological irregularities is steadily growing. These include proteins with circular permutations, segment-swapping, context-dependent folding or chameleon sequences that can adopt alternative secondary structures. Their non-trivial structural relationships are readily identified during expert analysis but their automatic identification using the existing computational tools still remains difficult or impossible. Such non-trivial cases of protein relationships are known to pose a problem to multiple alignment algorithms and to impede comparative modeling studies. They support a new emerging concept of evolutionary changeable protein fold, which creates practical difficulties for the hierarchical classifications of protein structures.To facilitate the understanding of, and to provide a comprehensive annotation of proteins with such non-trivial structural relationships we have created SISYPHUS ([Sigmaomeganuphiomicronzeta]--in Greek crafty), a compendium to the SCOP database. The SISYPHUS database contains a collection of manually curated structural alignments and their inter-relationships. The multiple alignments are constructed for protein structural regions that range from oligomeric biological units, or individual domains to fragments of different size. The SISYPHUS multiple alignments are displayed with SPICE, a browser that provides an integrated view of protein sequences, structures and their annotations. The database is available from http://sisyphus.mrc-cpe.cam.ac.uk.
随着结构数据量的不断增加,具有拓扑不规则性的同源蛋白质结构的数量正在稳步增长。这些包括具有环形排列、片段交换、上下文依赖折叠或能采用替代二级结构的变色龙序列的蛋白质。它们复杂的结构关系在专家分析过程中很容易识别,但使用现有的计算工具自动识别它们仍然很困难甚至不可能。已知这种复杂的蛋白质关系情况会给多序列比对算法带来问题,并阻碍比较建模研究。它们支持一种新出现的进化可变蛋白质折叠概念,这给蛋白质结构的层次分类带来了实际困难。为了便于理解并对具有这种复杂结构关系的蛋白质进行全面注释,我们创建了西西弗斯数据库(SISYPHUS,在希腊语中意为“狡猾的”),它是SCOP数据库的一个补充。西西弗斯数据库包含一组人工整理的结构比对及其相互关系。多序列比对是针对从寡聚生物单元、单个结构域到不同大小片段的蛋白质结构区域构建的。西西弗斯多序列比对通过SPICE浏览器显示,该浏览器提供了蛋白质序列、结构及其注释的综合视图。该数据库可从http://sisyphus.mrc-cpe.cam.ac.uk获取。