School of Computing and Engineering, University of Missouri at Kansas City, Kansas City, United States of America.
PLoS One. 2019 Mar 15;14(3):e0213712. doi: 10.1371/journal.pone.0213712. eCollection 2019.
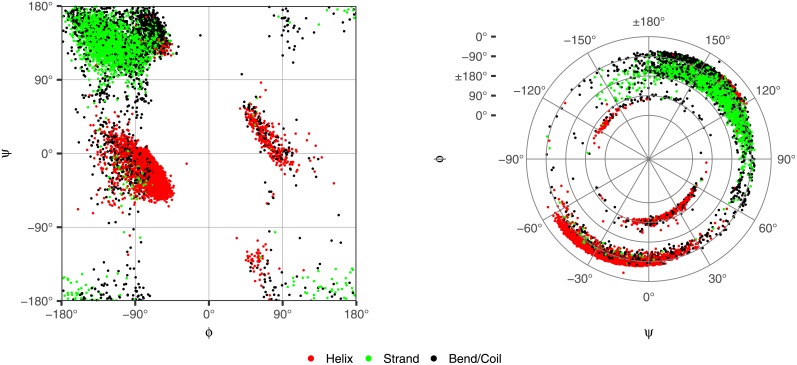
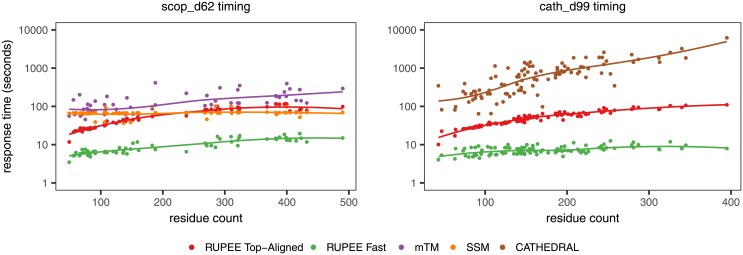
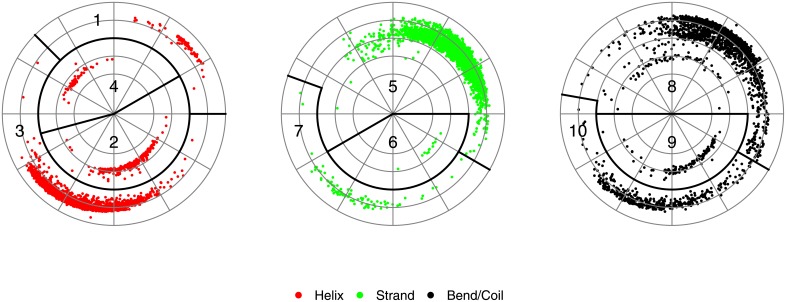
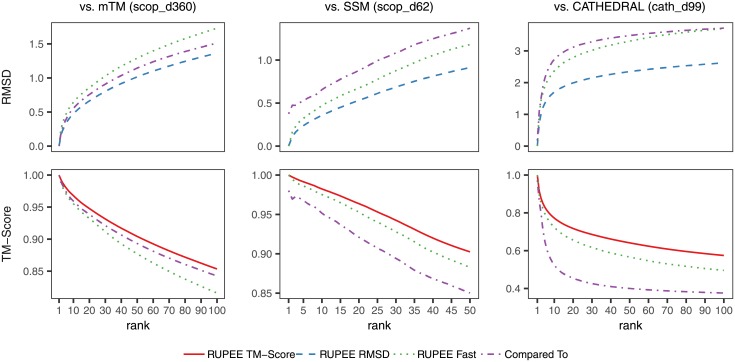
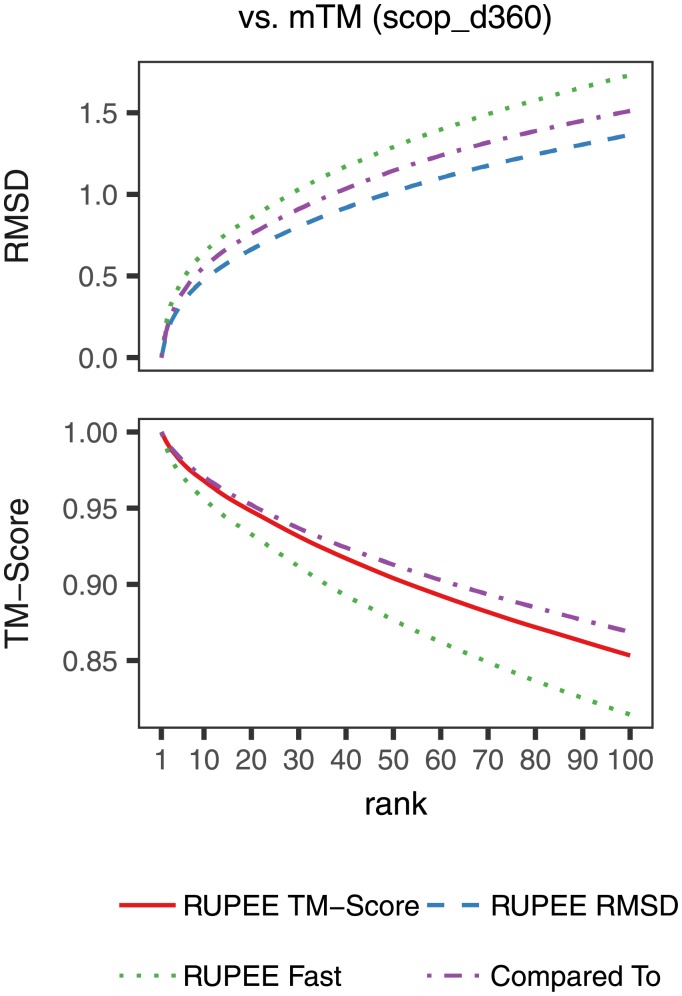
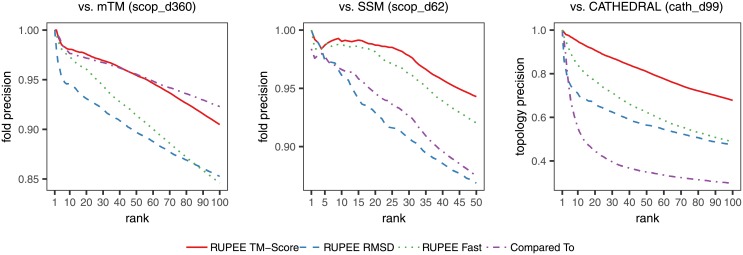
Given the close relationship between protein structure and function, protein structure searches have long played an established role in bioinformatics. Despite their maturity, existing protein structure searches either use simplifying assumptions or compromise between fast response times and quality of results. These limitations can prevent the easy and efficient exploration of relationships between protein structures, which is the norm in other areas of inquiry. To address these limitations we have developed RUPEE, a fast and accurate purely geometric structure search combining techniques from information retrieval and big data with a novel approach to encoding sequences of torsion angles. Comparing our results to the output of mTM, SSM, and the CATHEDRAL structural scan, it is clear that RUPEE has set a new bar for purely geometric big data approaches to protein structure searches. RUPEE in top-aligned mode produces equal or better results than the best available protein structure searches, and RUPEE in fast mode demonstrates the fastest response times coupled with high quality results. The RUPEE protein structure search is available at https://ayoubresearch.com. Code and data are available at https://github.com/rayoub/rupee.
鉴于蛋白质结构与功能之间的密切关系,蛋白质结构搜索在生物信息学中一直发挥着重要作用。尽管它们已经很成熟,但现有的蛋白质结构搜索要么使用简化的假设,要么在快速响应时间和结果质量之间做出妥协。这些限制可能会阻碍对蛋白质结构之间关系的轻松和高效探索,而这在其他研究领域是很常见的。为了解决这些限制,我们开发了 RUPEE,这是一种快速而准确的纯几何结构搜索,结合了信息检索和大数据技术,以及一种对扭转角序列进行编码的新方法。将我们的结果与 mTM、SSM 和 CATHEDRAL 结构扫描的输出进行比较,可以清楚地看出,RUPEE 为蛋白质结构搜索的纯几何大数据方法设定了一个新的标杆。RUPEE 在最高对齐模式下产生的结果与最好的可用蛋白质结构搜索相同或更好,而 RUPEE 在快速模式下则展示了最快的响应时间和高质量的结果。RUPEE 蛋白质结构搜索可在 https://ayoubresearch.com 上使用。代码和数据可在 https://github.com/rayoub/rupee 上获取。