Cao Feng, Zhou Tong, Simpson Dennis, Zhou Yingchun, Boyer Jayne, Chen Bo, Jin Taiyi, Cordeiro-Stone Marila, Kaufmann William
Department of Toxicology, School of Public Health, Medical Center of Fudan University, Shanghai, China.
Toxicol Appl Pharmacol. 2007 Jan 15;218(2):174-85. doi: 10.1016/j.taap.2006.10.031. Epub 2006 Nov 11.
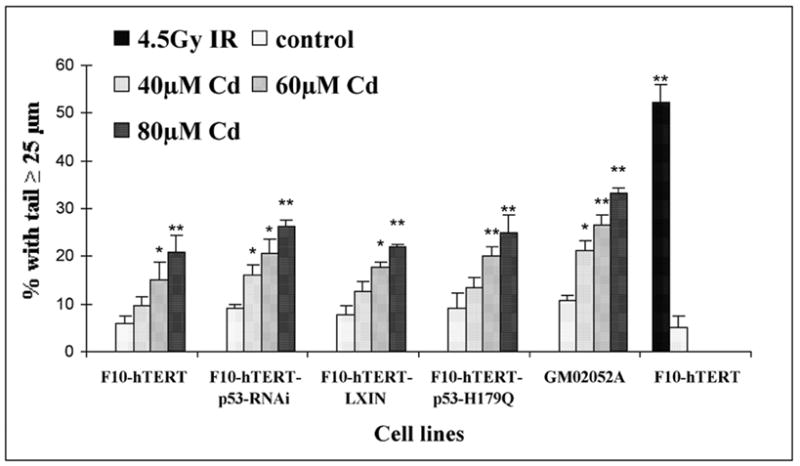
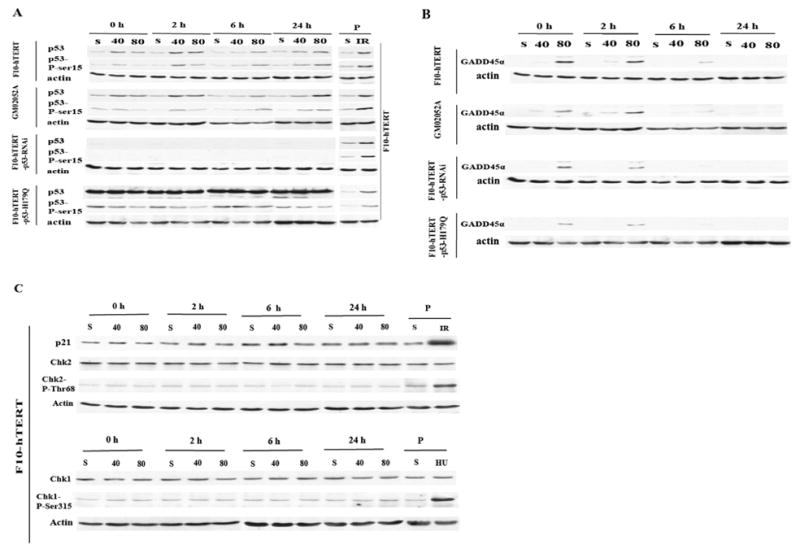
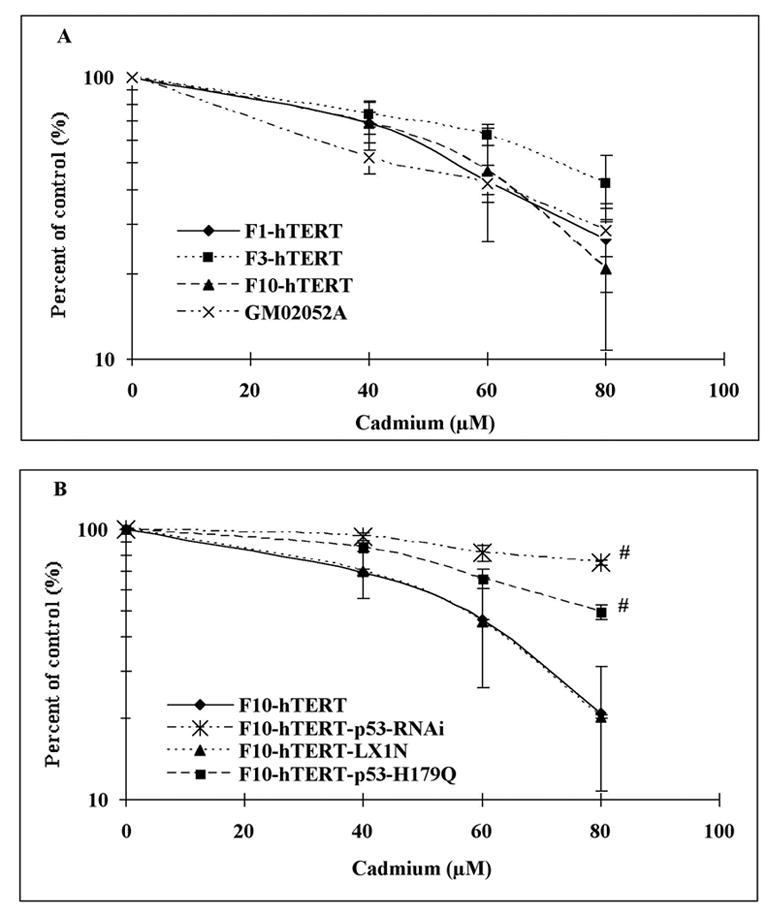
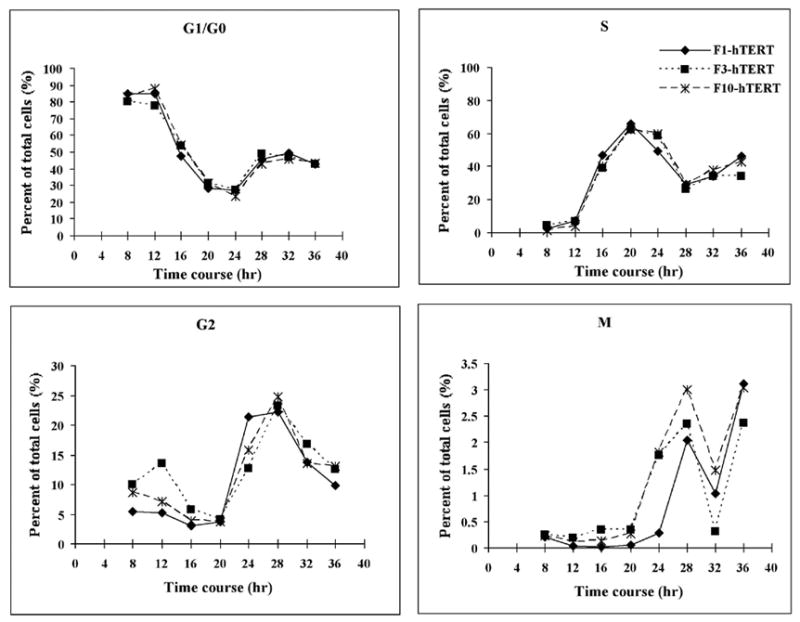
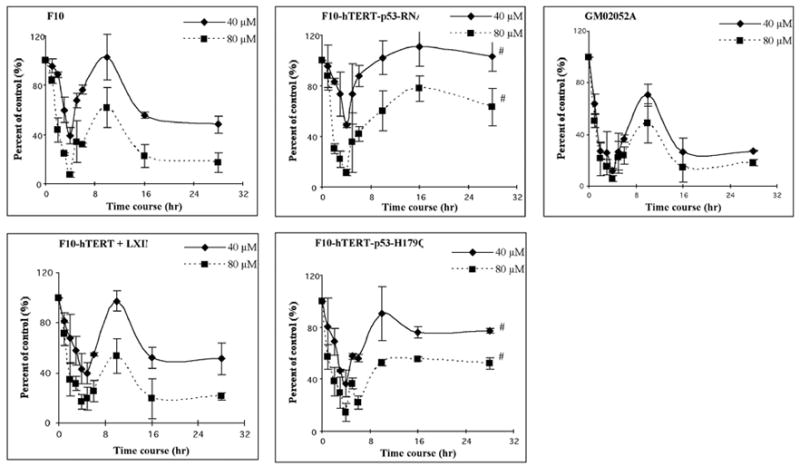
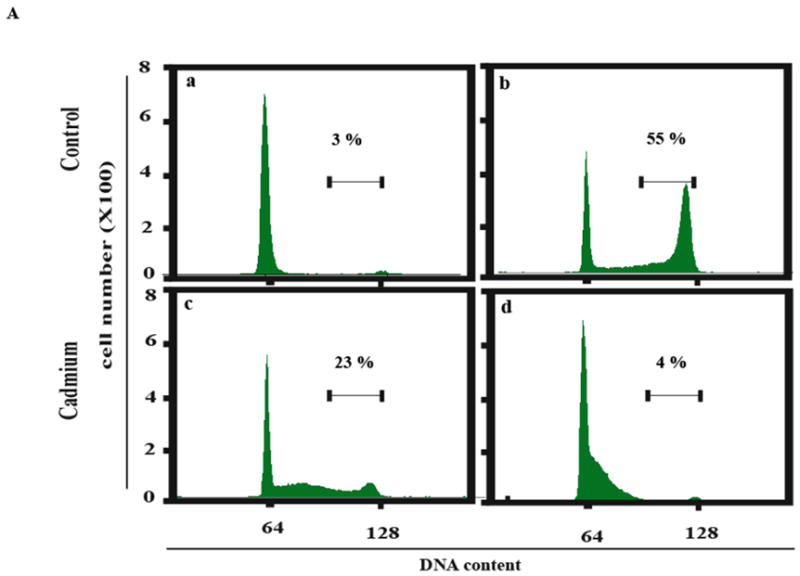
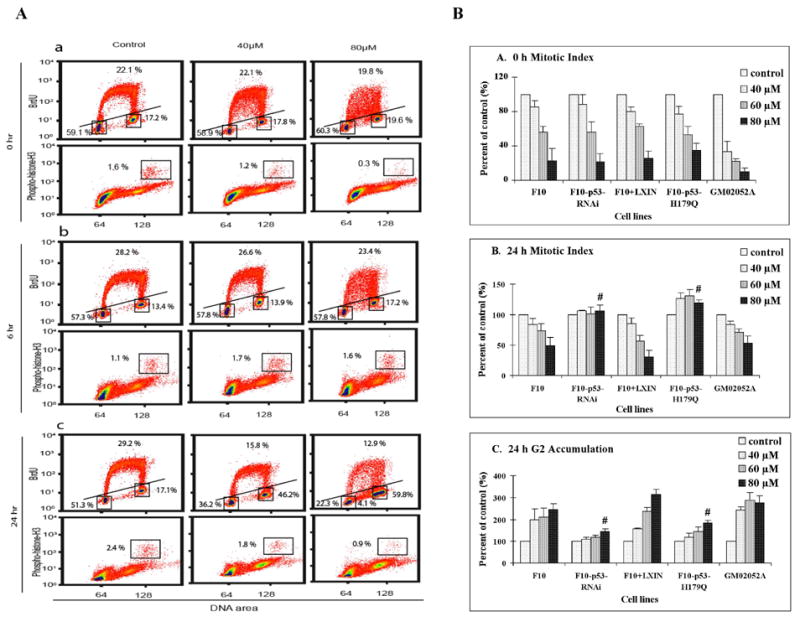
This study focused on the activation of cell cycle checkpoint responses in diploid human fibroblasts that were treated with cadmium chloride and the potential roles of ATM and p53 signaling pathways in cadmium-induced responses. The alkaline comet assay indicated that cadmium caused a dose-dependent increase in DNA damage. Cells that were rendered p53-defective by expression of a dominant-negative p53 allele or knockdown of p53 mRNA were more resistant to cadmium-induced inactivation of colony formation than normal and ataxia telangiectasia (AT) cells. Synchronized fibroblasts in S were more sensitive to cadmium toxicity than cells in G1, suggesting that cadmium may target some element of DNA replication. Cadmium produced a dose- and time-dependent inhibition of DNA synthesis. An immediate inhibition was associated with severe delay in progression through S phase and a delayed inhibition seen 24 h after treatment was associated with accumulation of cells in G2. AT and normal cells displayed similar patterns of inhibition of DNA synthesis and G2 delay after treatment with cadmium, while p53-defective cells displayed significantly less of the delayed inhibition of DNA synthesis and accumulation in G2 post-treatment. Total p53 protein and ser15-phosphorylated p53 were induced by cadmium in normal and AT cells. The p53 transactivation target Gadd45alpha was induced in both p53-effective and p53-defective cells after 4 h cadmium treatment, and this was associated with an acute inhibition of mitosis. Cadmium produced a very unusual pattern of toxicity in human fibroblasts, inhibiting DNA replication and inducing p53-dependent growth arrest but without induction of p21(Cip1/Waf1) or activation of Chk1.
本研究聚焦于用氯化镉处理的二倍体人成纤维细胞中细胞周期检查点反应的激活,以及ATM和p53信号通路在镉诱导反应中的潜在作用。碱性彗星试验表明,镉导致DNA损伤呈剂量依赖性增加。通过表达显性负性p53等位基因或敲低p53 mRNA而使p53功能缺陷的细胞,比正常细胞和共济失调毛细血管扩张症(AT)细胞对镉诱导的集落形成失活更具抗性。处于S期的同步化成纤维细胞比处于G1期的细胞对镉毒性更敏感,这表明镉可能靶向DNA复制的某些元件。镉产生剂量和时间依赖性的DNA合成抑制。即时抑制与S期进程的严重延迟相关,而处理后24小时出现的延迟抑制与细胞在G2期的积累相关。AT细胞和正常细胞在用镉处理后显示出相似的DNA合成抑制模式和G2期延迟,而p53功能缺陷的细胞在处理后显示出明显较少的DNA合成延迟抑制和G2期积累。正常细胞和AT细胞中的总p53蛋白和ser15磷酸化的p53由镉诱导产生。镉处理4小时后,p53转录激活靶点Gadd45α在p53功能有效和功能缺陷的细胞中均被诱导,这与有丝分裂的急性抑制相关。镉在人成纤维细胞中产生了一种非常不寻常的毒性模式,抑制DNA复制并诱导p53依赖性生长停滞,但不诱导p21(Cip1/Waf1)或激活Chk1。