Delbos Frédéric, Aoufouchi Said, Faili Ahmad, Weill Jean-Claude, Reynaud Claude-Agnès
Institut National de la Santé et de la Recherche Médicale U783 (Développement du système immunitaire) and Université Paris René Descartes, Faculté de Médecine René Descartes, Site Necker-Enfants Malades, 75730 Paris Cedex 15, France.
J Exp Med. 2007 Jan 22;204(1):17-23. doi: 10.1084/jem.20062131. Epub 2006 Dec 26.
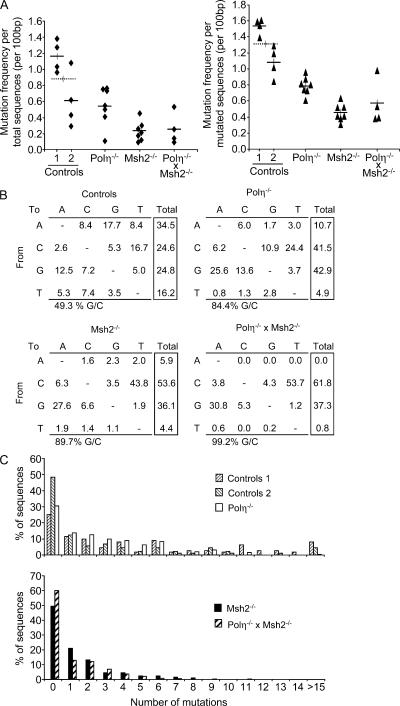
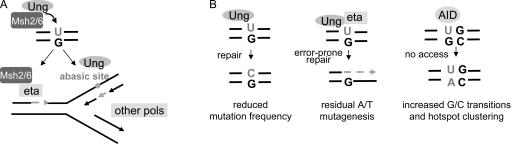
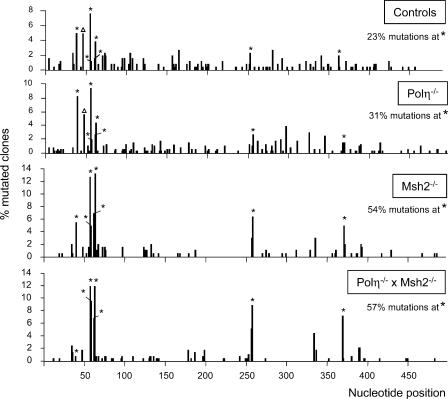
Mutations at A/T bases within immunoglobulin genes have been shown to be generated by a repair pathway involving the DNA-binding moiety of the mismatch repair complex constituted by the MSH2-MSH6 proteins, together with DNA polymerase eta (pol eta). However, residual A/T mutagenesis is still observed upon inactivation in the mouse of each of these factors, suggesting that the panel of activities involved might be more complex. We reported previously (Delbos, F., A. De Smet, A. Faili, S. Aoufouchi, J.-C. Weill, and C.-A. Reynaud. 2005. J. Exp. Med. 201:1191-1196) that residual A/T mutagenesis in pol eta-deficient mice was likely contributed by another enzyme not normally involved in hypermutation, DNA polymerase kappa, which is mobilized in the absence of the normal polymerase partner. We report the complete absence of A/T mutations in MSH2-pol eta double-deficient mice, thus indicating that the residual A/T mutagenesis in MSH2-deficient mice is contributed by pol eta, now recruited by uracil N-glycosylase, the second DNA repair pathway involved in hypermutation. We propose that this particular recruitment of pol eta corresponds to a profound modification of the function of uracil glycosylase in the absence of the mismatch repair complex, suggesting that MSH2-MSH6 actively prevent uracil glycosylase from error-free repair during hypermutation. pol eta thus appears to be the sole contributor of A/T mutations in the normal physiological context.
免疫球蛋白基因内A/T碱基处的突变已被证明是由一种修复途径产生的,该途径涉及由MSH2 - MSH6蛋白与DNA聚合酶η(pol η)构成的错配修复复合物的DNA结合部分。然而,在这些因子中的每一个在小鼠体内失活后,仍可观察到残留的A/T诱变现象,这表明所涉及的活性组合可能更为复杂。我们之前报道过(德尔博斯,F.,A. 德·斯梅特,A. 法伊利,S. 奥富奇,J.-C. 韦伊,以及C.-A. 雷诺。2005年。《实验医学杂志》201:1191 - 1196),在缺乏pol η的小鼠中残留的A/T诱变可能是由另一种通常不参与高突变的酶——DNA聚合酶κ导致的,该酶在正常聚合酶伴侣缺失的情况下被动员起来。我们报告在MSH2 - pol η双缺陷小鼠中完全不存在A/T突变,因此表明在缺乏MSH2的小鼠中残留的A/T诱变是由pol η导致的,现在pol η由尿嘧啶N - 糖基化酶招募,尿嘧啶N - 糖基化酶是参与高突变的第二条DNA修复途径。我们提出,在错配修复复合物缺失的情况下,pol η的这种特殊招募对应于尿嘧啶糖基化酶功能的深刻改变,这表明MSH2 - MSH6在高突变过程中积极阻止尿嘧啶糖基化酶进行无差错修复。因此,在正常生理环境中,pol η似乎是A/T突变的唯一贡献者。