Pournara Iosifina, Wernisch Lorenz
School of Crystallography, Birkbeck College, University of London, London, UK.
BMC Bioinformatics. 2007 Feb 23;8:61. doi: 10.1186/1471-2105-8-61.
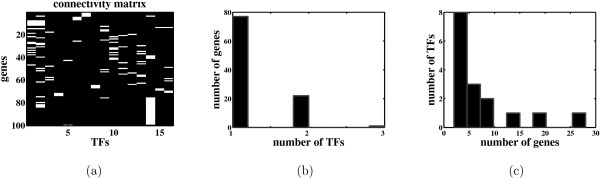
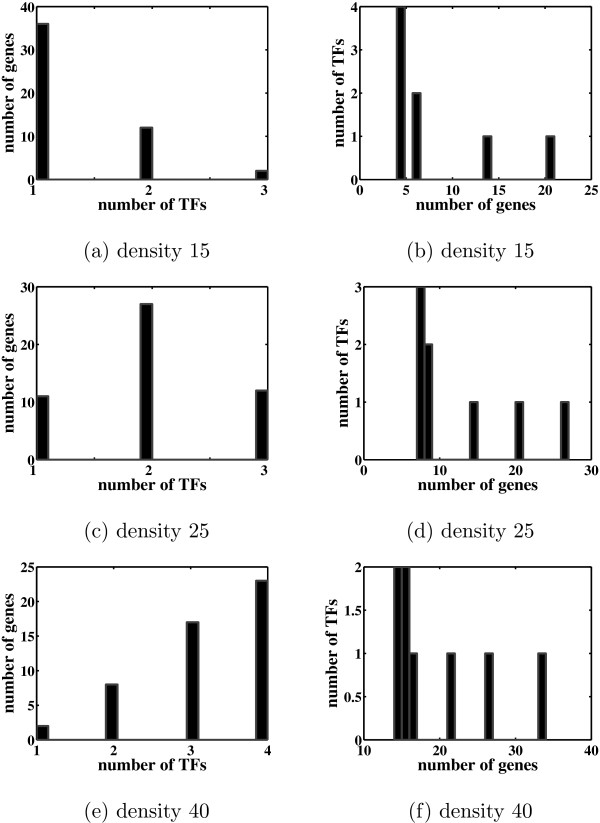
Most existing algorithms for the inference of the structure of gene regulatory networks from gene expression data assume that the activity levels of transcription factors (TFs) are proportional to their mRNA levels. This assumption is invalid for most biological systems. However, one might be able to reconstruct unobserved activity profiles of TFs from the expression profiles of target genes. A simple model is a two-layer network with unobserved TF variables in the first layer and observed gene expression variables in the second layer. TFs are connected to regulated genes by weighted edges. The weights, known as factor loadings, indicate the strength and direction of regulation. Of particular interest are methods that produce sparse networks, networks with few edges, since it is known that most genes are regulated by only a small number of TFs, and most TFs regulate only a small number of genes.
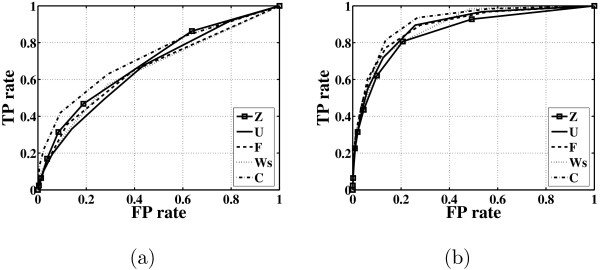
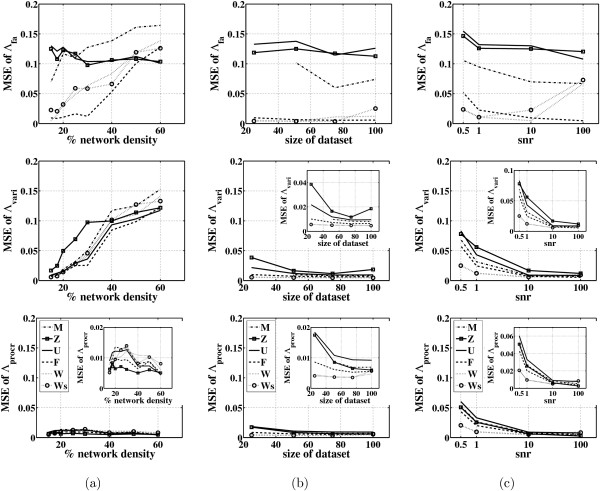
In this paper, we explore the performance of five factor analysis algorithms, Bayesian as well as classical, on problems with biological context using both simulated and real data. Factor analysis (FA) models are used in order to describe a larger number of observed variables by a smaller number of unobserved variables, the factors, whereby all correlation between observed variables is explained by common factors. Bayesian FA methods allow one to infer sparse networks by enforcing sparsity through priors. In contrast, in the classical FA, matrix rotation methods are used to enforce sparsity and thus to increase the interpretability of the inferred factor loadings matrix. However, we also show that Bayesian FA models that do not impose sparsity through the priors can still be used for the reconstruction of a gene regulatory network if applied in conjunction with matrix rotation methods. Finally, we show the added advantage of merging the information derived from all algorithms in order to obtain a combined result.
Most of the algorithms tested are successful in reconstructing the connectivity structure as well as the TF profiles. Moreover, we demonstrate that if the underlying network is sparse it is still possible to reconstruct hidden activity profiles of TFs to some degree without prior connectivity information.
大多数现有的从基因表达数据推断基因调控网络结构的算法都假定转录因子(TFs)的活性水平与其mRNA水平成正比。这一假设对大多数生物系统而言是无效的。然而,人们或许能够从靶基因的表达谱中重建未观测到的TFs活性谱。一个简单的模型是一个两层网络,第一层有未观测到的TF变量,第二层有观测到的基因表达变量。TFs通过加权边与受调控基因相连。这些权重,即因子负荷,表明调控的强度和方向。特别令人感兴趣的是能产生稀疏网络(边很少的网络)的方法,因为已知大多数基因仅受少数TFs调控,且大多数TFs仅调控少数基因。
在本文中,我们使用模拟数据和真实数据,探讨了五种因子分析算法(贝叶斯算法和经典算法)在具有生物学背景的问题上的性能。因子分析(FA)模型用于通过较少数量的未观测变量(即因子)来描述大量观测变量,从而使观测变量之间的所有相关性都由公共因子来解释。贝叶斯FA方法允许通过先验来强制稀疏性,从而推断稀疏网络。相比之下,在经典FA中,使用矩阵旋转方法来强制稀疏性,进而提高推断出的因子负荷矩阵的可解释性。然而,我们还表明,如果将不通过先验强制稀疏性的贝叶斯FA模型与矩阵旋转方法结合应用,仍然可用于基因调控网络的重建。最后,我们展示了合并所有算法得出的信息以获得综合结果的额外优势。
大多数测试算法在重建连接结构以及TFs谱方面都取得了成功。此外,我们证明,如果基础网络是稀疏的,那么在没有先验连接信息的情况下,仍然有可能在一定程度上重建TFs的隐藏活性谱。