Department of Computer Science, University of Sheffield, Sheffield, UK.
Amazon Research, Cambridge, UK.
Bioinformatics. 2017 Dec 1;33(23):3776-3783. doi: 10.1093/bioinformatics/btx508.
Regulation of gene expression in prokaryotes involves complex co-regulatory mechanisms involving large numbers of transcriptional regulatory proteins and their target genes. Uncovering these genome-scale interactions constitutes a major bottleneck in systems biology. Sparse latent factor models, assuming activity of transcription factors (TFs) as unobserved, provide a biologically interpretable modelling framework, integrating gene expression and genome-wide binding data, but at the same time pose a hard computational inference problem. Existing probabilistic inference methods for such models rely on subjective filtering and suffer from scalability issues, thus are not well-suited for realistic genome-scale applications.
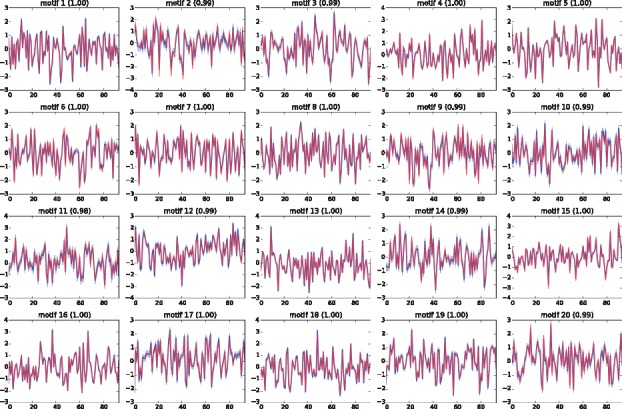
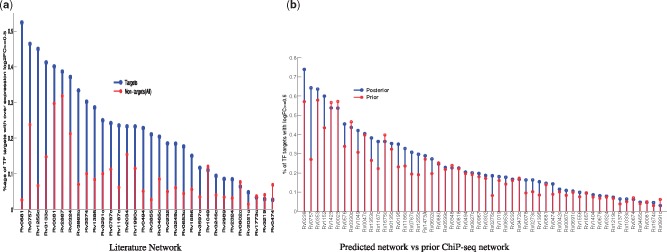
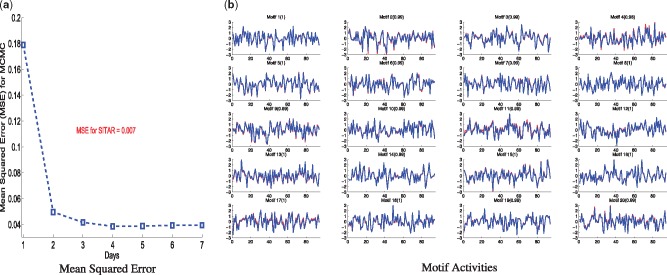
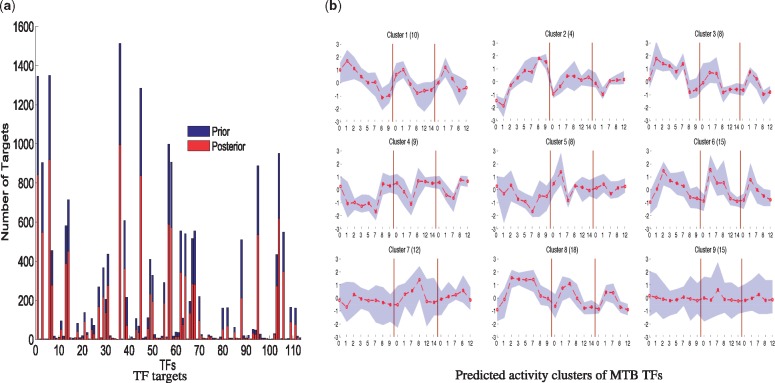
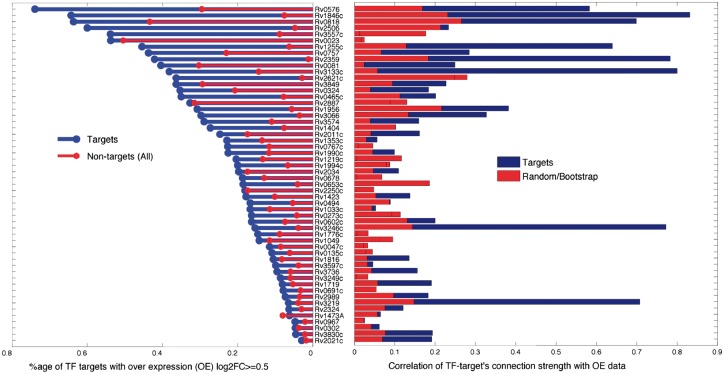
We present a fast Bayesian sparse factor model, which takes input gene expression and binding sites data, either from ChIP-seq experiments or motif predictions, and outputs active TF-gene links as well as latent TF activities. Our method employs an efficient variational Bayes scheme for model inference enabling its application to large datasets which was not feasible with existing MCMC-based inference methods for such models. We validate our method on synthetic data against a similar model in the literature, employing MCMC for inference, and obtain comparable results with a small fraction of the computational time. We also apply our method to large-scale data from Mycobacterium tuberculosis involving ChIP-seq data on 113 TFs and matched gene expression data for 3863 putative target genes. We evaluate our predictions using an independent transcriptomics experiment involving over-expression of TFs.
An easy-to-use Jupyter notebook demo of our method with data is available at https://github.com/zhenwendai/SITAR.
mudassar.iqbal@manchester.ac.uk.
Supplementary data are available at Bioinformatics online.
原核生物中基因表达的调控涉及复杂的协同调控机制,涉及大量转录调控蛋白及其靶基因。揭示这些基因组规模的相互作用是系统生物学的一个主要瓶颈。稀疏潜在因子模型,假设转录因子(TFs)的活性是不可观测的,为基因表达和全基因组结合数据提供了一个具有生物学可解释性的建模框架,但同时也构成了一个困难的计算推断问题。现有的此类模型的概率推断方法依赖于主观过滤,并且存在可扩展性问题,因此不适用于现实的基因组规模应用。
我们提出了一种快速贝叶斯稀疏因子模型,它接受来自 ChIP-seq 实验或基序预测的基因表达和结合位点数据作为输入,并输出活跃的 TF-基因链接以及潜在的 TF 活性。我们的方法采用了一种有效的变分贝叶斯方案进行模型推断,使其能够应用于大型数据集,而这在以前基于 MCMC 的此类模型的推断方法中是不可行的。我们在合成数据上对我们的方法进行了验证,与文献中的类似模型进行了比较,使用 MCMC 进行推断,并在计算时间的一小部分内获得了可比的结果。我们还将我们的方法应用于涉及 113 个 TF 的 ChIP-seq 数据和 3863 个假定靶基因的匹配基因表达数据的大型结核分枝杆菌数据集。我们使用涉及 TF 过表达的独立转录组学实验来评估我们的预测。
我们的方法的带有数据的易于使用的 Jupyter 笔记本演示可在 https://github.com/zhenwendai/SITAR 上获得。
mudassar.iqbal@manchester.ac.uk。
补充数据可在 Bioinformatics 在线获得。