Rockwell Nathan C, Lagarias J Clark
Section of Molecular and Cellular Biology, University of California, Davis, California 95616, USA.
BMC Bioinformatics. 2007 Apr 11;8:123. doi: 10.1186/1471-2105-8-123.
Over the past decade, a number of tools have emerged for the examination of homology relationships among protein sequences in a structural context. Most recent software implementations for such analysis are tied to specific molecular viewing programs, which can be problematic for collaborations involving multiple viewing environments. Incorporation into larger packages also adds complications for users interested in adding their own scoring schemes or in analyzing proteins incorporating unusual amino acid residues such as selenocysteine.
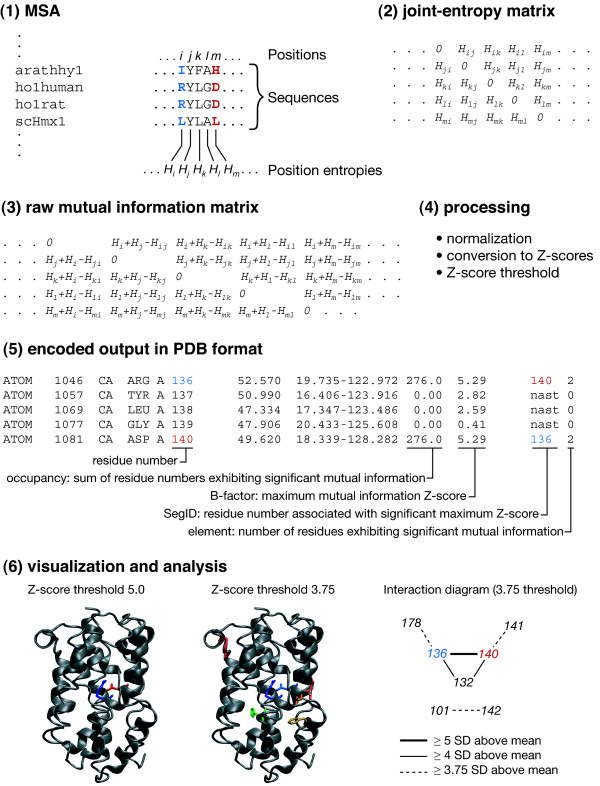
We describe homolmapper, a command-line application for mapping information from a multiple protein sequence alignment onto a protein structure for analysis in the viewing software of the user's choice. Homolmapper is small (under 250 K for the application itself) and is written in Python to ensure portability. It is released for non-commercial use under a modified University of California BSD license. Homolmapper permits facile import of additional scoring schemes and can incorporate arbitrary additional amino acids to allow handling of residues such as selenocysteine or pyrrolysine. Homolmapper also provides tools for defining and analyzing subfamilies relative to a larger alignment, for mutual information analysis, and for rapidly visualizing the locations of mutations and multi-residue motifs.
Homolmapper is a useful tool for analysis of homology relationships among proteins in a structural context. There is also extensive, example-driven documentation available. More information about homolmapper is available at http://www.mcb.ucdavis.edu/faculty-labs/lagarias/homolmapper_home/homolmapper%20web%20page.htm.
在过去十年中,出现了许多用于在结构背景下检查蛋白质序列间同源关系的工具。此类分析的最新软件实现与特定的分子可视化程序相关联,这对于涉及多种可视化环境的合作可能会有问题。纳入更大的软件包也给有兴趣添加自己评分方案或分析包含不寻常氨基酸残基(如硒代半胱氨酸)的蛋白质的用户带来了复杂性。
我们描述了homolmapper,这是一个命令行应用程序,用于将来自多序列蛋白质比对的信息映射到蛋白质结构上,以便在用户选择的可视化软件中进行分析。homolmapper体积小(应用程序本身不到250K),用Python编写以确保可移植性。它在修改后的加利福尼亚大学伯克利分校BSD许可下发布供非商业使用。homolmapper允许轻松导入额外的评分方案,并可纳入任意额外的氨基酸,以处理诸如硒代半胱氨酸或吡咯赖氨酸之类的残基。homolmapper还提供了用于相对于更大比对定义和分析亚家族、进行互信息分析以及快速可视化突变和多残基基序位置的工具。
homolmapper是用于在结构背景下分析蛋白质同源关系的有用工具。还有大量的、以示例为驱动的文档。有关homolmapper的更多信息可在http://www.mcb.ucdavis.edu/faculty-labs/lagarias/homolmapper_home/homolmapper%20web%20page.htm获取。