Gaile Daniel P, Miecznikowski Jeffrey C
Department of Biostatistics, University at Buffalo, Buffalo, New York, USA.
BMC Genomics. 2007 Apr 19;8:105. doi: 10.1186/1471-2164-8-105.
We provide a re-analysis of the Golden Spike dataset, a first generation "spike-in" control microarray dataset. The original analysis of the Golden Spike dataset was presented in a manuscript by Choe et al. and raised questions concerning the performance of several statistical methods for the control of the false discovery rate (across a set of tests for differential expression). These original findings are now in question as it has been reported that the p-values associated with the tests of differential expression for null probesets (i.e., probesets designed to be fold change 1 across the two arms of the experiment) are not uniformly distributed. Two recent publications have speculated as to the reasons the null distributions are non-uniform. A publication by Dabney and Storey concludes that the non-uniform distributions of null p-values are the direct consequence of an experimental design which requires technical replicates to approximate biological replicates. Irizarry et al. identify four characteristics of the feature level data (three related to experimental design and one artifact). Irizarry et al. argue that the four observed characteristics imply that the assumptions common to most pre-processing algorithms are not satisfied and hence the expression measure methodologies considered by Choe et al. are likely to be flawed.
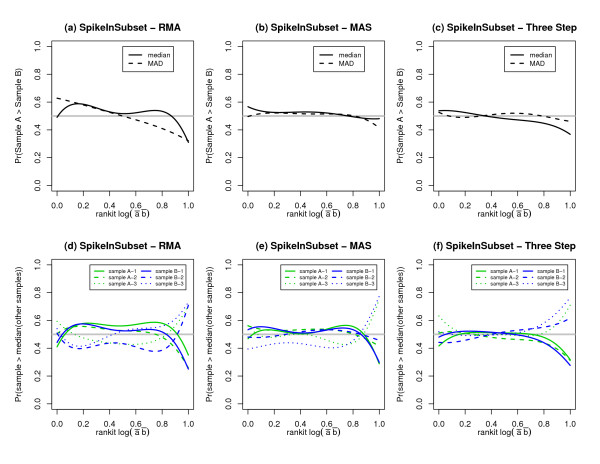
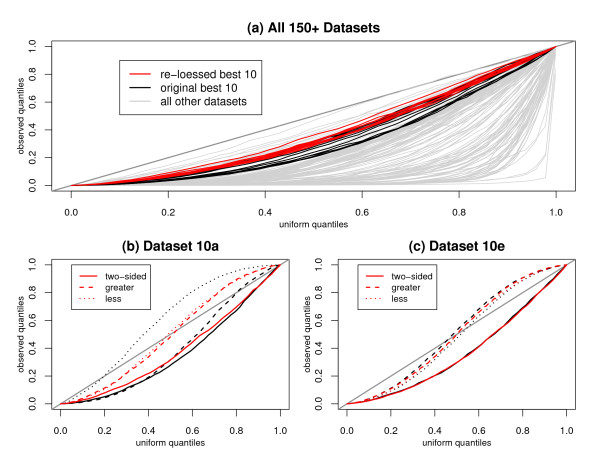
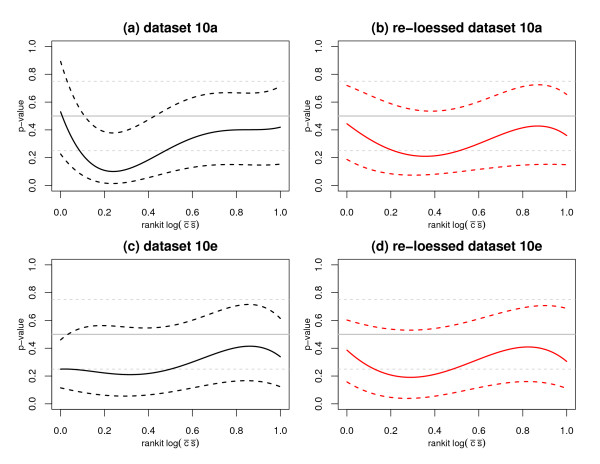
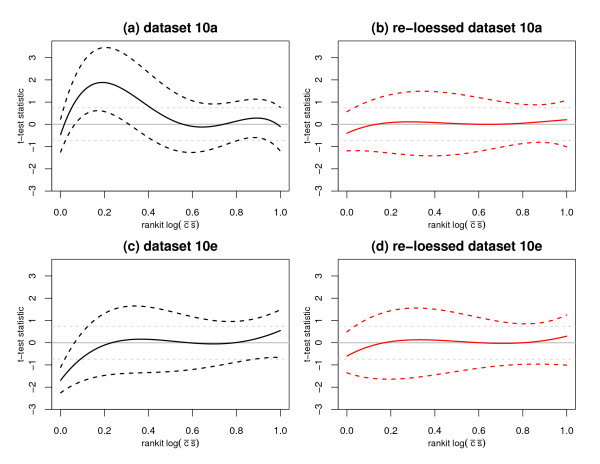
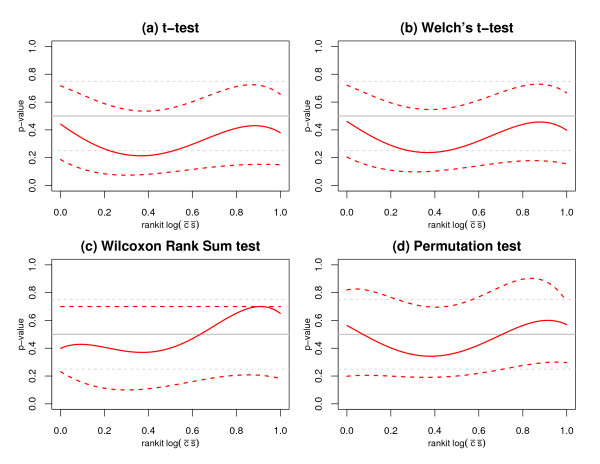
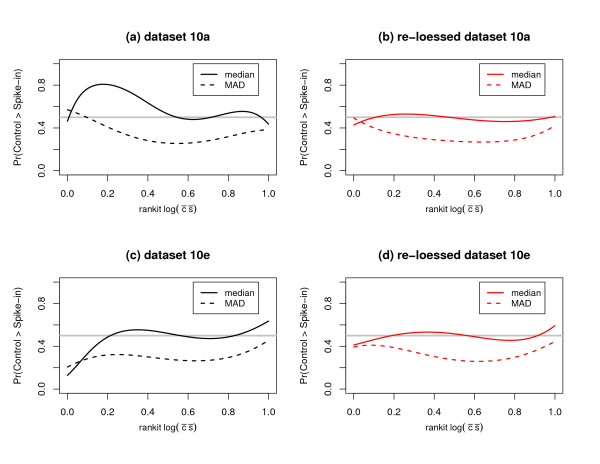
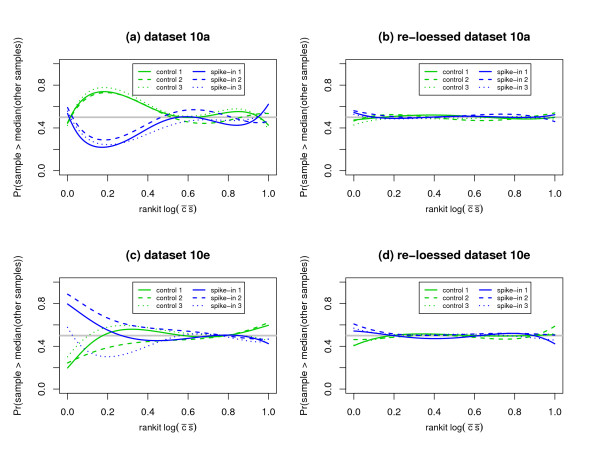
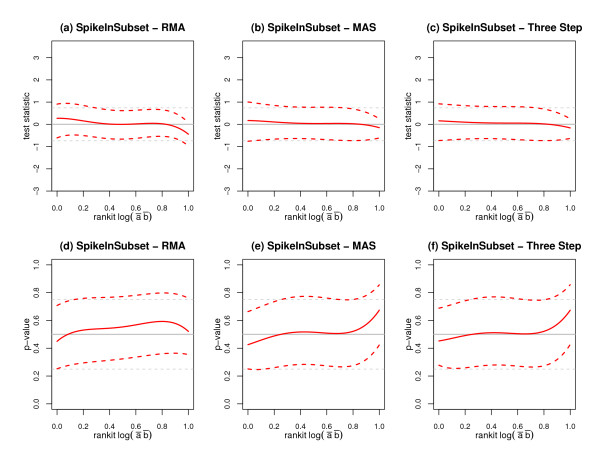
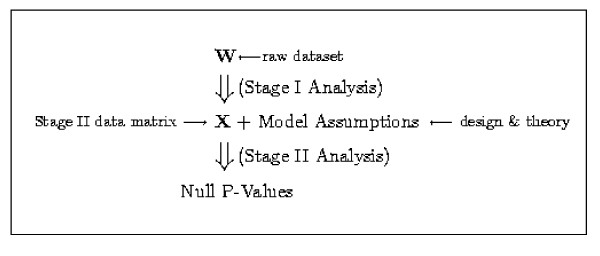
We replicate and extend the analyses of Dabney and Storey and present our results in the context of a two stage analysis. We provide evidence that the Stage I pre-processing algorithms considered in Dabney and Storey fail to provide expression values that are adequately centered or scaled. Furthermore, we demonstrate that the distributions of the p-values, test statistics, and probabilities associated with the relative locations and variabilities of the Stage II expression values vary with signal intensity. We provide diagnostic plots and a simple logistic regression based test statistic to detect these intensity related defects in the processed data.
We agree with Dabney and Storey that the null p-values considered in Choe et al. are indeed non-uniform. We also agree with the conclusion that, given current pre-processing technologies, the Golden Spike dataset should not serve as a reference dataset to evaluate false discovery rate controlling methodologies. However, we disagree with the assessment that the non-uniform p-values are merely the byproduct of testing for differential expression under the incorrect assumption that chip data are approximate to biological replicates. Whereas Dabney and Storey attribute the non-uniform p-values to violations of the Stage II model assumptions, we provide evidence that the non-uniformity can be attributed to the failure of the Stage I analyses to correct for systematic biases in the raw data matrix. Although we do not speculate as to the root cause of these systematic biases, the observations made in Irizarry et al. appear to be consistent with our findings. Whereas Irizarry et al. describe the effect of the experimental design on the feature level data, we consider the effect on the underlying multivariate distribution of putative null p-values. We demonstrate that the putative null distributions corresponding to the pre-processing algorithms considered in Choe et al. are all intensity dependent. This dependence serves to invalidate statistical inference based upon standard two sample test statistics. We identify a flaw in the characterization of the appropriate "null" probesets described in Choe et al. and we provide a corrected analysis which reduces (but does not eliminate) the intensity dependent effects.
我们对金标准数据集进行了重新分析,这是一个第一代“掺入式”对照微阵列数据集。Choe等人的一篇论文中展示了对金标准数据集的原始分析,并对几种用于控制错误发现率(在一组差异表达测试中)的统计方法的性能提出了疑问。由于有报道称与无效探针集(即设计为在实验的两个臂上的倍数变化为1的探针集)的差异表达测试相关的p值并非均匀分布,这些原始发现现在受到了质疑。最近的两篇论文推测了无效分布不均匀的原因。Dabney和Storey的一篇论文得出结论,无效p值的非均匀分布是实验设计的直接结果,该实验设计需要技术重复来近似生物学重复。Irizarry等人确定了特征水平数据的四个特征(三个与实验设计有关,一个是人为因素)。Irizarry等人认为观察到的这四个特征意味着大多数预处理算法共有的假设不成立,因此Choe等人考虑的表达测量方法可能存在缺陷。
我们重复并扩展了Dabney和Storey的分析,并在两阶段分析的背景下展示了我们的结果。我们提供证据表明,Dabney和Storey中考虑的第一阶段预处理算法未能提供充分居中或缩放的表达值。此外,我们证明与第二阶段表达值的相对位置和变异性相关的p值、检验统计量和概率的分布随信号强度而变化。我们提供诊断图和一个基于简单逻辑回归的检验统计量,以检测处理后数据中这些与强度相关的缺陷。
我们同意Dabney和Storey的观点,即Choe等人中考虑的无效p值确实不均匀。我们也同意这样的结论,即鉴于当前的预处理技术,金标准数据集不应作为评估错误发现率控制方法的参考数据集。然而,我们不同意那种认为非均匀p值仅仅是在芯片数据近似于生物学重复这一错误假设下进行差异表达测试的副产品的评估。虽然Dabney和Storey将非均匀p值归因于违反第二阶段模型假设,但我们提供证据表明,这种不均匀性可归因于第一阶段分析未能校正原始数据矩阵中的系统偏差。尽管我们没有推测这些系统偏差的根本原因,但Irizarry等人的观察结果似乎与我们的发现一致。Irizarry等人描述了实验设计对特征水平数据的影响,而我们考虑的是对假定无效p值的潜在多元分布的影响。我们证明,Choe等人中考虑的预处理算法对应的假定无效分布都与强度有关。这种依赖性使得基于标准双样本检验统计量的统计推断无效。我们发现了Choe等人中描述的适当“无效”探针集特征的一个缺陷,并提供了一种校正分析,该分析减少了(但没有消除)与强度相关的影响。