National Cancer Institute Center for Bioinformatics, Rockville, Maryland, United States of America.
PLoS One. 2007 May 9;2(5):e425. doi: 10.1371/journal.pone.0000425.
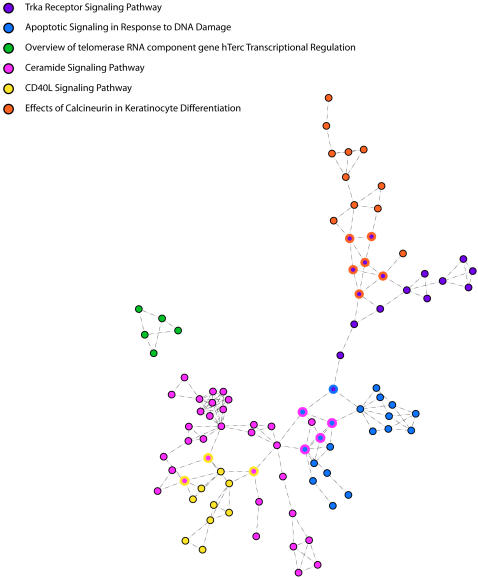
Cancer is recognized to be a family of gene-based diseases whose causes are to be found in disruptions of basic biologic processes. An increasingly deep catalogue of canonical networks details the specific molecular interaction of genes and their products. However, mapping of disease phenotypes to alterations of these networks of interactions is accomplished indirectly and non-systematically. Here we objectively identify pathways associated with malignancy, staging, and outcome in cancer through application of an analytic approach that systematically evaluates differences in the activity and consistency of interactions within canonical biologic processes. Using large collections of publicly accessible genome-wide gene expression, we identify small, common sets of pathways - Trka Receptor, Apoptosis response to DNA Damage, Ceramide, Telomerase, CD40L and Calcineurin - whose differences robustly distinguish diverse tumor types from corresponding normal samples, predict tumor grade, and distinguish phenotypes such as estrogen receptor status and p53 mutation state. Pathways identified through this analysis perform as well or better than phenotypes used in the original studies in predicting cancer outcome. This approach provides a means to use genome-wide characterizations to map key biological processes to important clinical features in disease.
癌症被认为是一种基于基因的疾病家族,其病因可归因于基本生物过程的中断。越来越多的规范网络目录详细说明了基因及其产物的特定分子相互作用。然而,将疾病表型映射到这些相互作用网络的改变是间接和非系统地完成的。在这里,我们通过应用一种分析方法客观地识别与恶性肿瘤、分期和癌症结果相关的途径,该方法系统地评估了规范生物过程中相互作用的活性和一致性的差异。我们使用大量公开的全基因组基因表达数据集,确定了小的、常见的途径集 - Trka 受体、DNA 损伤的细胞凋亡反应、神经酰胺、端粒酶、CD40L 和钙调神经磷酸酶 - 这些途径集的差异可以可靠地区分不同的肿瘤类型与相应的正常样本,预测肿瘤分级,并区分雌激素受体状态和 p53 突变状态等表型。通过这种分析识别的途径在预测癌症结果方面的表现与原始研究中使用的表型一样好或更好。这种方法提供了一种利用全基因组特征将关键生物学过程映射到疾病重要临床特征的方法。