Liu Shu-Qun, Meng Zhao-Hui, Yang Jin-Kui, Fu Yun-Xin, Zhang Ke-Qin
Laboratory for Conservation and Utilization of Bio-resources, Yunnan University, Yunnan, Kunming, PR China.
BMC Struct Biol. 2007 May 18;7:33. doi: 10.1186/1472-6807-7-33.
Serine proteases secreted by nematode and insect pathogenic fungi are bio-control agents which have commercial potential for developing into effective bio-pesticides. A thorough understanding of the structural and functional features of these proteases would significantly assist with targeting the design of efficient bio-control agents.
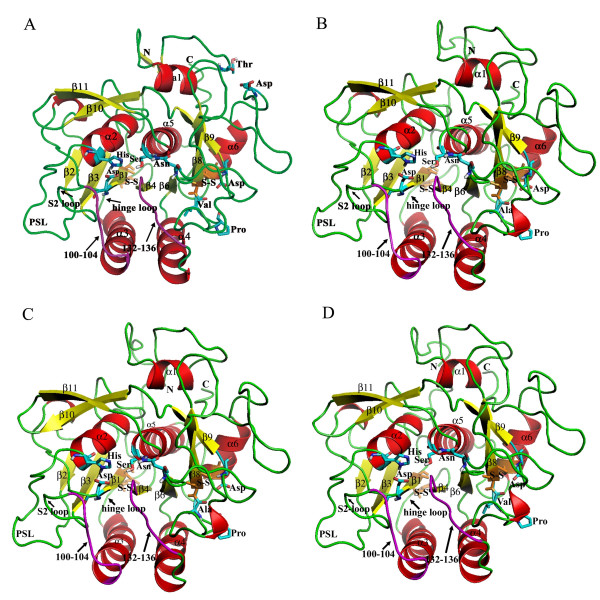
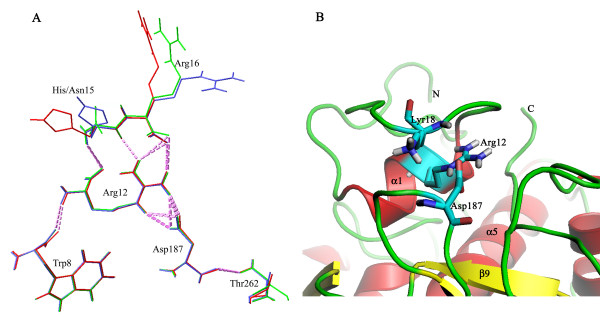
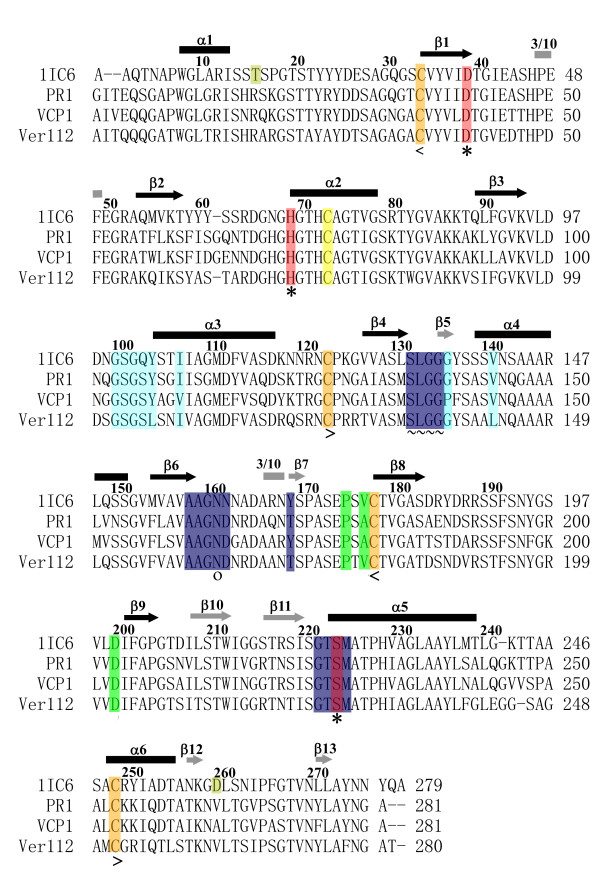
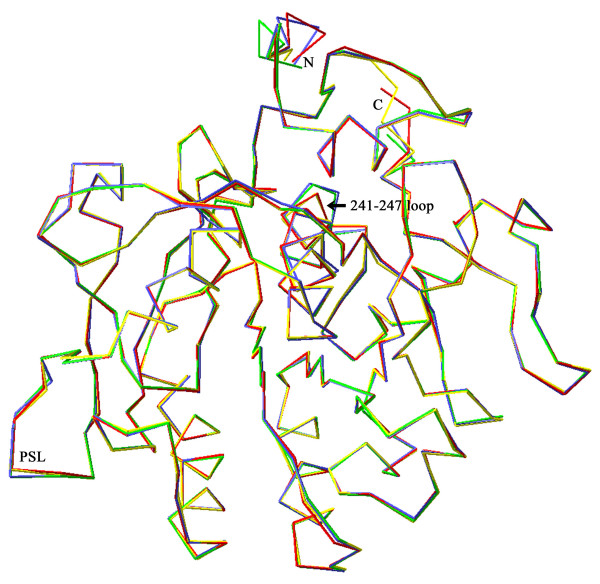
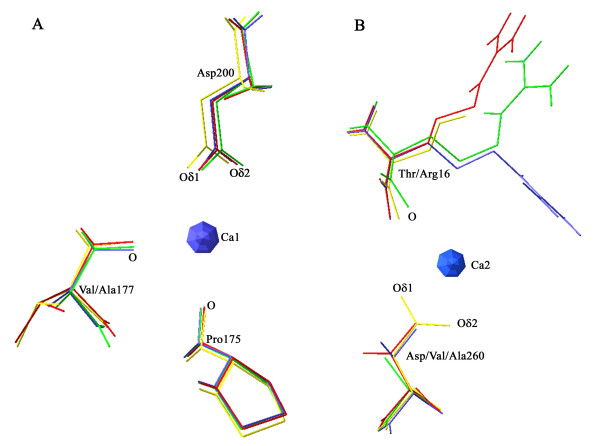
Structural models of serine proteases PR1 from entomophagous fungus, Ver112 and VCP1 from nematophagous fungi, have been modeled using the homology modeling technique based on the crystal coordinate of the proteinase K. In combination with multiple sequence alignment, these models suggest one similar calcium-binding site and two common disulfide bridges in the three cuticle-degrading enzymes. In addition, the predicted models of the three cuticle-degrading enzymes present an essentially identical backbone topology and similar geometric properties with the exception of a limited number of sites exhibiting relatively large local conformational differences only in some surface loops and the N-, C termini. However, they differ from each other in the electrostatic surface potential, in hydrophobicity and size of the S4 substrate-binding pocket, and in the number and distribution of hydrogen bonds and salt bridges within regions that are part of or in close proximity to the S2-loop.
These differences likely lead to variations in substrate specificity and catalytic efficiency among the three enzymes. Amino acid polymorphisms in cuticle-degrading enzymes were discussed with respect to functional effects and host preference. It is hoped that these structural models would provide a further basis for exploitation of these serine proteases from pathogenic fungi as effective bio-control agents.
线虫和昆虫病原真菌分泌的丝氨酸蛋白酶是生物防治剂,具有开发成有效生物农药的商业潜力。深入了解这些蛋白酶的结构和功能特征将极大地有助于高效生物防治剂的靶向设计。
利用基于蛋白酶K晶体坐标的同源建模技术,对食虫真菌PR1、线虫寄生真菌Ver112和VCP1的丝氨酸蛋白酶进行了结构建模。结合多序列比对,这些模型表明这三种角质层降解酶中有一个相似的钙结合位点和两个共同的二硫键。此外,这三种角质层降解酶的预测模型呈现出基本相同的主链拓扑结构和相似的几何性质,只是在一些表面环以及N、C末端中,只有有限数量的位点表现出相对较大的局部构象差异。然而,它们在静电表面电位、S4底物结合口袋的疏水性和大小、以及S2环附近或其一部分区域内氢键和盐桥的数量和分布方面存在差异。
这些差异可能导致这三种酶在底物特异性和催化效率上有所不同。针对功能效应和宿主偏好,讨论了角质层降解酶中的氨基酸多态性。希望这些结构模型能为将这些病原真菌中的丝氨酸蛋白酶开发成有效的生物防治剂提供进一步的依据。