Butland Stefanie L, Devon Rebecca S, Huang Yong, Mead Carri-Lyn, Meynert Alison M, Neal Scott J, Lee Soo Sen, Wilkinson Anna, Yang George S, Yuen Macaire M S, Hayden Michael R, Holt Robert A, Leavitt Blair R, Ouellette B F Francis
UBC Bioinformatics Centre, Michael Smith Laboratories, University of British Columbia, Vancouver, Canada.
BMC Genomics. 2007 May 22;8:126. doi: 10.1186/1471-2164-8-126.
Expansion of polyglutamine-encoding CAG trinucleotide repeats has been identified as the pathogenic mutation in nine different genes associated with neurodegenerative disorders. The majority of individuals clinically diagnosed with spinocerebellar ataxia do not have mutations within known disease genes, and it is likely that additional ataxias or Huntington disease-like disorders will be found to be caused by this common mutational mechanism. We set out to determine the length distributions of CAG-polyglutamine tracts for the entire human genome in a set of healthy individuals in order to characterize the nature of polyglutamine repeat length variation across the human genome, to establish the background against which pathogenic repeat expansions can be detected, and to prioritize candidate genes for repeat expansion disorders.
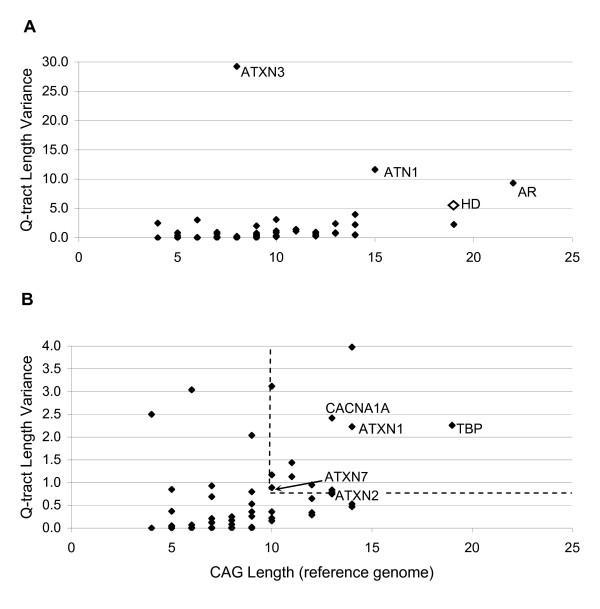
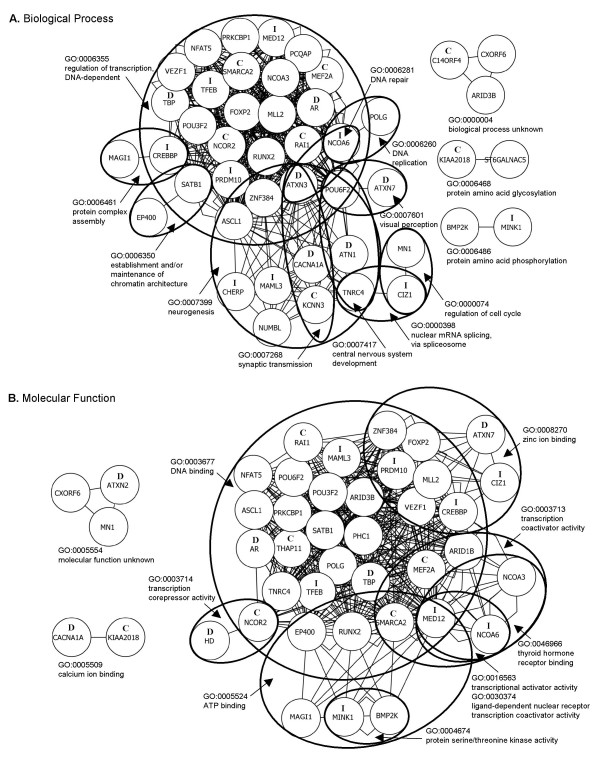
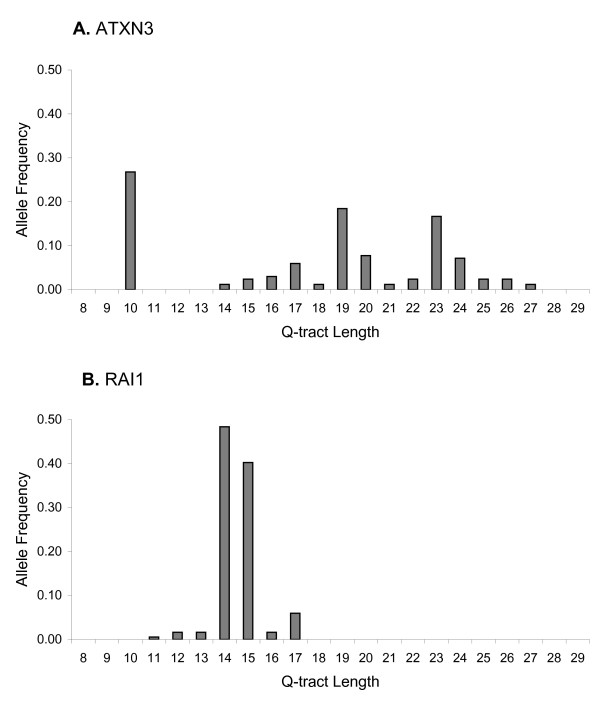
We found that repeats, including those in known disease genes, have unique distributions of glutamine tract lengths, as measured by fragment analysis of PCR-amplified repeat regions. This emphasizes the need to characterize each distribution and avoid making generalizations between loci. The best predictors of known disease genes were occurrence of a long CAG-tract uninterrupted by CAA codons in their reference genome sequence, and high glutamine tract length variance in the normal population. We used these parameters to identify eight priority candidate genes for polyglutamine expansion disorders. Twelve CAG-polyglutamine repeats were invariant and these can likely be excluded as candidates. We outline some confusion in the literature about this type of data, difficulties in comparing such data between publications, and its application to studies of disease prevalence in different populations. Analysis of Gene Ontology-based functions of CAG-polyglutamine-containing genes provided a visual framework for interpretation of these genes' functions. All nine known disease genes were involved in DNA-dependent regulation of transcription or in neurogenesis, as were all of the well-characterized priority candidate genes.
This publication makes freely available the normal distributions of CAG-polyglutamine repeats in the human genome. Using these background distributions, against which pathogenic expansions can be identified, we have begun screening for mutations in individuals clinically diagnosed with novel forms of spinocerebellar ataxia or Huntington disease-like disorders who do not have identified mutations within the known disease-associated genes.
编码聚谷氨酰胺的CAG三核苷酸重复序列的扩增已被确定为与神经退行性疾病相关的9种不同基因中的致病突变。大多数临床诊断为脊髓小脑共济失调的个体在已知疾病基因中没有突变,很可能会发现其他共济失调或亨廷顿病样疾病是由这种常见的突变机制引起的。我们着手确定一组健康个体中整个人类基因组CAG - 聚谷氨酰胺序列的长度分布,以表征整个人类基因组中聚谷氨酰胺重复长度变异的性质,建立可检测致病重复序列扩增的背景,并对重复序列扩增疾病的候选基因进行优先级排序。
我们发现,通过对PCR扩增的重复区域进行片段分析测量,包括已知疾病基因中的重复序列在内,重复序列具有独特的谷氨酰胺序列长度分布。这强调了表征每种分布并避免在不同位点之间进行一概而论的必要性。已知疾病基因的最佳预测指标是其参考基因组序列中存在未被CAA密码子打断的长CAG序列,以及正常人群中谷氨酰胺序列长度的高变异性。我们使用这些参数确定了8个聚谷氨酰胺扩增疾病的优先候选基因。12个CAG - 聚谷氨酰胺重复序列是不变的,这些很可能可以排除在候选基因之外。我们概述了文献中关于此类数据的一些混淆、不同出版物之间比较此类数据的困难以及其在不同人群疾病患病率研究中的应用。对含CAG - 聚谷氨酰胺基因基于基因本体论的功能分析为解释这些基因的功能提供了一个直观的框架。所有9个已知疾病基因都参与了DNA依赖性转录调控或神经发生,所有特征明确的优先候选基因也是如此。
本出版物免费提供了人类基因组中CAG - 聚谷氨酰胺重复序列的正常分布。利用这些背景分布可以识别致病扩增,我们已经开始对临床诊断为新型脊髓小脑共济失调或亨廷顿病样疾病但在已知疾病相关基因中未发现突变的个体进行突变筛查。