Rose Tobias, Efendic Suad, Rupnik Marjan
European Neuroscience Institute-Göttingen, 37073 Göttingen, Germany.
J Gen Physiol. 2007 Jun;129(6):493-508. doi: 10.1085/jgp.200609604.
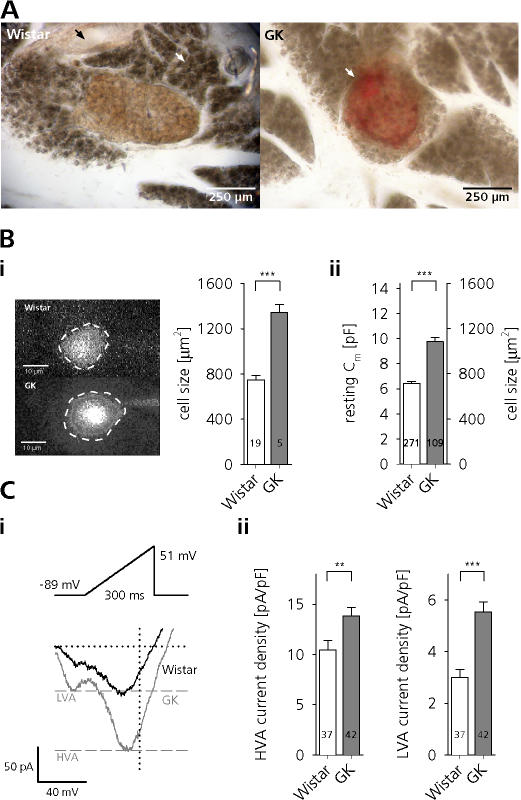
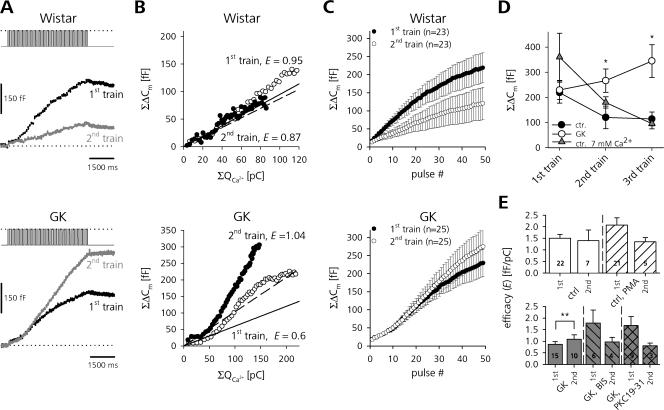
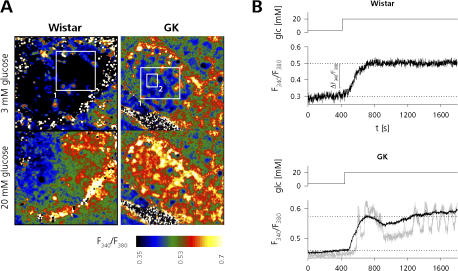
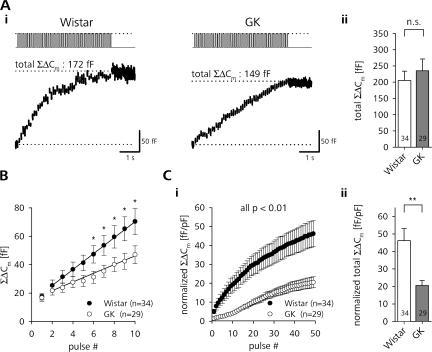
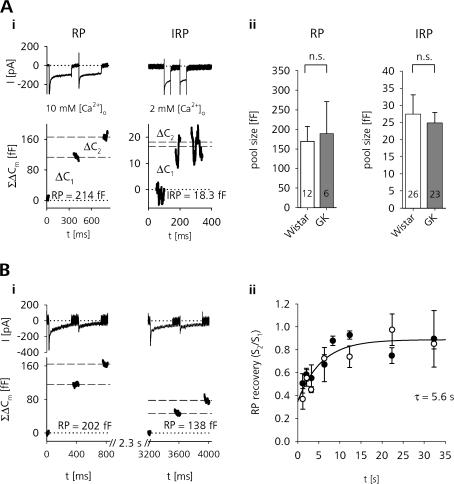
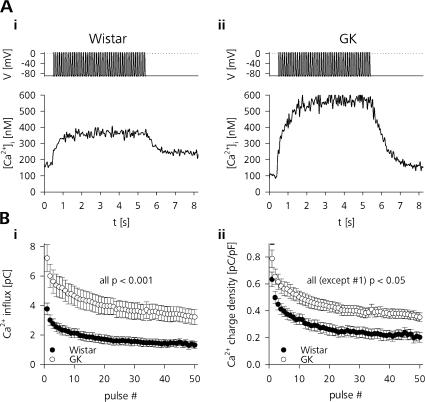
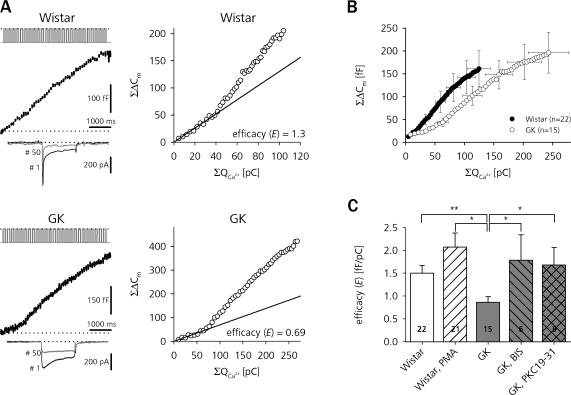
The Goto Kakizaki (GK) rat is a widely used animal model to study defective glucose-stimulated insulin release in type-2 diabetes (T2D). As in T2D patients, the expression of several proteins involved in Ca(2+)-dependent exocytosis of insulin-containing large dense-core vesicles is dysregulated in this model. So far, a defect in late steps of insulin secretion could not be demonstrated. To resolve this apparent contradiction, we studied Ca(2+)-secretion coupling of healthy and GK rat beta cells in acute pancreatic tissue slices by assessing exocytosis with high time-resolution membrane capacitance measurements. We found that beta cells of GK rats respond to glucose stimulation with a normal increase in the cytosolic Ca(2+) concentration. During trains of depolarizing pulses, the secretory activity from GK rat beta cells was defective in spite of upregulated cell size and doubled voltage-activated Ca(2+) currents. In GK rat beta cells, evoked Ca(2+) entry was significantly less efficient in triggering release than in nondiabetic controls. This impairment was neither due to a decrease of functional vesicle pool sizes nor due to different kinetics of pool refilling. Strong stimulation with two successive trains of depolarizing pulses led to a prominent activity-dependent facilitation of release in GK rat beta cells, whereas secretion in controls was unaffected. Broad-spectrum inhibition of PKC sensitized Ca(2+)-dependent exocytosis, whereas it prevented the activity-dependent facilitation in GK rat beta cells. We conclude that a decrease in the sensitivity of the GK rat beta-cell to depolarization-evoked Ca(2+) influx is involved in defective glucose-stimulated insulin secretion. Furthermore, we discuss a role for constitutively increased activity of one or more PKC isoenzymes in diabetic rat beta cells.
Goto Kakizaki(GK)大鼠是一种广泛用于研究2型糖尿病(T2D)中葡萄糖刺激的胰岛素释放缺陷的动物模型。与T2D患者一样,在该模型中,参与含胰岛素的大致密核心囊泡的Ca(2 +)依赖性胞吐作用的几种蛋白质的表达失调。到目前为止,尚未证明胰岛素分泌后期存在缺陷。为了解决这一明显的矛盾,我们通过高时间分辨率膜电容测量评估胞吐作用,研究了急性胰腺组织切片中健康和GK大鼠β细胞的Ca(2 +)分泌偶联。我们发现,GK大鼠的β细胞对葡萄糖刺激的反应是胞质Ca(2 +)浓度正常增加。在去极化脉冲序列期间,尽管细胞大小上调且电压激活的Ca(2 +)电流增加了一倍,但GK大鼠β细胞的分泌活性仍存在缺陷。在GK大鼠β细胞中,诱发的Ca(2 +)内流在触发释放方面的效率明显低于非糖尿病对照组。这种损伤既不是由于功能性囊泡池大小的减少,也不是由于池再填充的不同动力学。用两列连续的去极化脉冲进行强刺激导致GK大鼠β细胞中释放的显著活性依赖性促进,而对照组中的分泌不受影响。蛋白激酶C(PKC)的广谱抑制使Ca(2 +)依赖性胞吐作用敏感,而它阻止了GK大鼠β细胞中的活性依赖性促进。我们得出结论,GK大鼠β细胞对去极化诱发的Ca(2 +)内流的敏感性降低与葡萄糖刺激的胰岛素分泌缺陷有关。此外,我们讨论了一种或多种PKC同工酶在糖尿病大鼠β细胞中组成性增加的活性的作用。