Ekins S, Mestres J, Testa B
ACT LLC, 1 Penn Plaza, New York, NY 10119, USA.
Br J Pharmacol. 2007 Sep;152(1):21-37. doi: 10.1038/sj.bjp.0707306. Epub 2007 Jun 4.
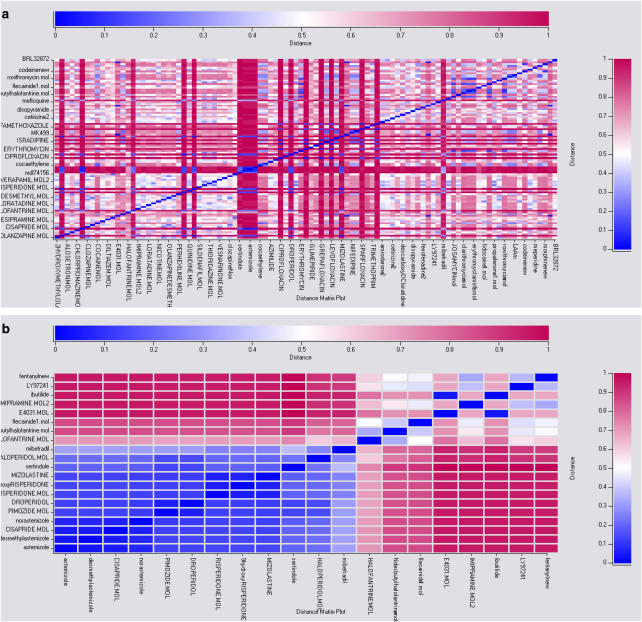
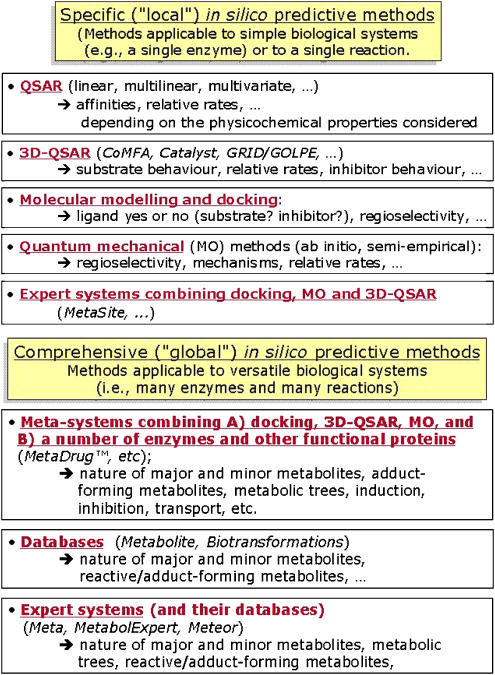
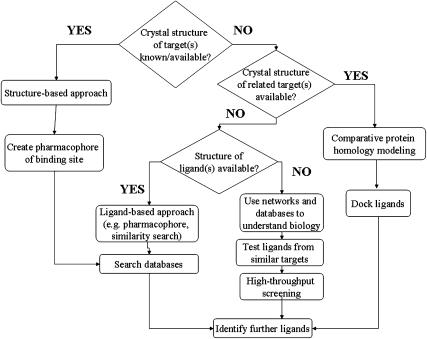
Computational (in silico) methods have been developed and widely applied to pharmacology hypothesis development and testing. These in silico methods include databases, quantitative structure-activity relationships, similarity searching, pharmacophores, homology models and other molecular modeling, machine learning, data mining, network analysis tools and data analysis tools that use a computer. Such methods have seen frequent use in the discovery and optimization of novel molecules with affinity to a target, the clarification of absorption, distribution, metabolism, excretion and toxicity properties as well as physicochemical characterization. The first part of this review discussed the methods that have been used for virtual ligand and target-based screening and profiling to predict biological activity. The aim of this second part of the review is to illustrate some of the varied applications of in silico methods for pharmacology in terms of the targets addressed. We will also discuss some of the advantages and disadvantages of in silico methods with respect to in vitro and in vivo methods for pharmacology research. Our conclusion is that the in silico pharmacology paradigm is ongoing and presents a rich array of opportunities that will assist in expediting the discovery of new targets, and ultimately lead to compounds with predicted biological activity for these novel targets.
计算(计算机模拟)方法已被开发并广泛应用于药理学假设的提出和验证。这些计算机模拟方法包括数据库、定量构效关系、相似性搜索、药效团、同源模型及其他分子模拟、机器学习、数据挖掘、网络分析工具以及使用计算机的数据分析工具。此类方法在发现和优化对靶点具有亲和力的新型分子、阐明吸收、分布、代谢、排泄和毒性特性以及物理化学表征方面经常被使用。本综述的第一部分讨论了用于虚拟配体和基于靶点的筛选及分析以预测生物活性的方法。本综述第二部分的目的是根据所涉及的靶点阐述计算机模拟方法在药理学中的一些不同应用。我们还将讨论计算机模拟方法相对于药理学研究的体外和体内方法的一些优缺点。我们的结论是,计算机模拟药理学范式仍在发展,并且提供了丰富的机会,将有助于加速新靶点的发现,并最终得到针对这些新靶点具有预测生物活性的化合物。