Hsu Chen-Ming, Chen Chien-Yu, Liu Baw-Jhiune, Huang Chih-Chang, Laio Min-Hung, Lin Chien-Chieh, Wu Tzung-Lin
Department of Computer Science and Engineering, Yuan Ze University, Chung-Li, Taiwan, ROC.
BMC Bioinformatics. 2007 May 24;8 Suppl 5(Suppl 5):S8. doi: 10.1186/1471-2105-8-S5-S8.
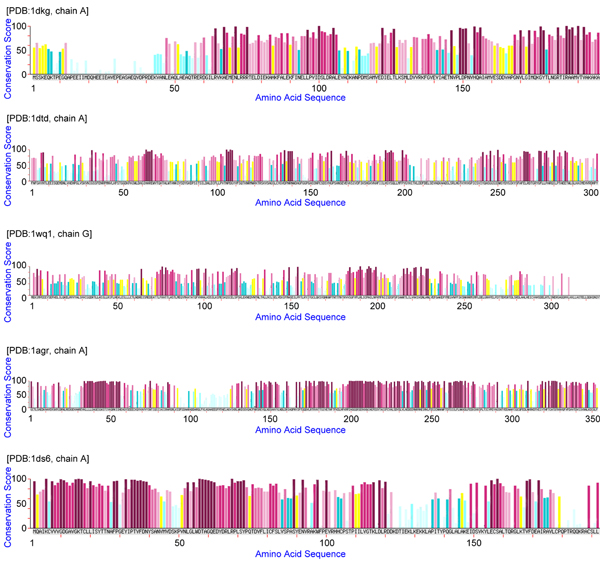
Identification of protein interacting sites is an important task in computational molecular biology. As more and more protein sequences are deposited without available structural information, it is strongly desirable to predict protein binding regions by their sequences alone. This paper presents a pattern mining approach to tackle this problem. It is observed that a functional region of protein structures usually consists of several peptide segments linked with large wildcard regions. Thus, the proposed mining technology considers large irregular gaps when growing patterns, in order to find the residues that are simultaneously conserved but largely separated on the sequences. A derived pattern is called a cluster-like pattern since the discovered conserved residues are always grouped into several blocks, which each corresponds to a local conserved region on the protein sequence.
The experiments conducted in this work demonstrate that the derived long patterns automatically discover the important residues that form one or several hot regions of protein-protein interactions. The methodology is evaluated by conducting experiments on the web server MAGIIC-PRO based on a well known benchmark containing 220 protein chains from 72 distinct complexes. Among the tested 218 proteins, there are 900 sequential blocks discovered, 4.25 blocks per protein chain on average. About 92% of the derived blocks are observed to be clustered in space with at least one of the other blocks, and about 66% of the blocks are found to be near the interface of protein-protein interactions. It is summarized that for about 83% of the tested proteins, at least two interacting blocks can be discovered by this approach.
This work aims to demonstrate that the important residues associated with the interface of protein-protein interactions may be automatically discovered by sequential pattern mining. The detected regions possess high conservation and thus are considered as the computational hot regions. This information would be useful to characterizing protein sequences, predicting protein function, finding potential partners, and facilitating protein docking for drug discovery.
识别蛋白质相互作用位点是计算分子生物学中的一项重要任务。随着越来越多的蛋白质序列在没有可用结构信息的情况下被存入数据库,仅通过序列来预测蛋白质结合区域的需求变得极为迫切。本文提出了一种模式挖掘方法来解决这一问题。据观察,蛋白质结构的功能区域通常由几个肽段与大的通配符区域相连组成。因此,所提出的挖掘技术在模式增长时考虑大的不规则间隙,以便找到在序列上同时保守但在很大程度上分隔开的残基。由于发现的保守残基总是被分组为几个块,每个块对应于蛋白质序列上的一个局部保守区域,所以导出的模式被称为类簇模式。
在这项工作中进行的实验表明,导出的长模式自动发现了形成蛋白质 - 蛋白质相互作用的一个或几个热点区域的重要残基。基于包含来自72个不同复合物的220条蛋白质链的著名基准,在网络服务器MAGIIC - PRO上进行实验对该方法进行了评估。在所测试的218种蛋白质中,共发现900个连续块,平均每条蛋白质链有4.25个块。观察到约92%的导出块在空间上与至少一个其他块聚集在一起,并且约66%的块位于蛋白质 - 蛋白质相互作用的界面附近。总结得出,对于约83%的测试蛋白质,通过这种方法至少可以发现两个相互作用块。
这项工作旨在证明通过序列模式挖掘可以自动发现与蛋白质 - 蛋白质相互作用界面相关的重要残基。检测到的区域具有高度保守性,因此被视为计算热点区域。这些信息对于表征蛋白质序列、预测蛋白质功能、寻找潜在伙伴以及促进用于药物发现的蛋白质对接将是有用的。