Lassmann Timo, Sonnhammer Erik Ll
Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden.
BMC Bioinformatics. 2007 May 24;8 Suppl 5(Suppl 5):S9. doi: 10.1186/1471-2105-8-S5-S9.
High quality multiple alignments are crucial in the transfer of annotation from one genome to another. Multiple alignment methods strive to achieve ever increasing levels of average accuracy on benchmark sets while the accuracy of individual alignments is often overlooked.
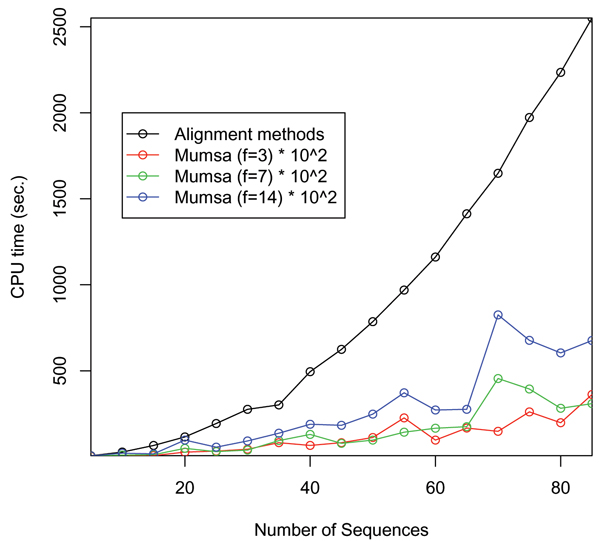
We have previously developed a method to automatically assess the accuracy and overall difficulty of multiple alignments. This was achieved by a per-residue comparison between alternate alignments of the same sequences. Here we present a key extension to this method, an algorithm to extract similarly aligned regions from several alignments and merge them into a new consensus alignment.
We demonstrate that the fraction of correctly aligned residues within the resulting alignments is increased by 25-100 percent compared to the original input alignments, as only the most reliably aligned parts are considered.
高质量的多序列比对在将注释从一个基因组转移到另一个基因组的过程中至关重要。多序列比对方法致力于在基准数据集上不断提高平均准确率,而单个比对的准确性常常被忽视。
我们之前开发了一种自动评估多序列比对的准确性和整体难度的方法。这是通过对相同序列的交替比对进行逐个残基比较来实现的。在此,我们展示了该方法的一个关键扩展,即一种从多个比对中提取相似比对区域并将它们合并成一个新的一致比对的算法。
我们证明,与原始输入比对相比,所得比对中正确比对残基的比例提高了25%至100%,因为只考虑了最可靠比对的部分。