Lassmann Timo, Sonnhammer Erik L L
Center for Genomics and Bioinformatics, Karolinska Institutet, Berzelius vag 35, S-17177 Stockholm, Sweden.
BMC Bioinformatics. 2005 Dec 12;6:298. doi: 10.1186/1471-2105-6-298.
The alignment of multiple protein sequences is a fundamental step in the analysis of biological data. It has traditionally been applied to analyzing protein families for conserved motifs, phylogeny, structural properties, and to improve sensitivity in homology searching. The availability of complete genome sequences has increased the demands on multiple sequence alignment (MSA) programs. Current MSA methods suffer from being either too inaccurate or too computationally expensive to be applied effectively in large-scale comparative genomics.
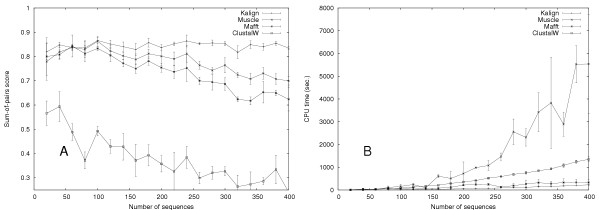
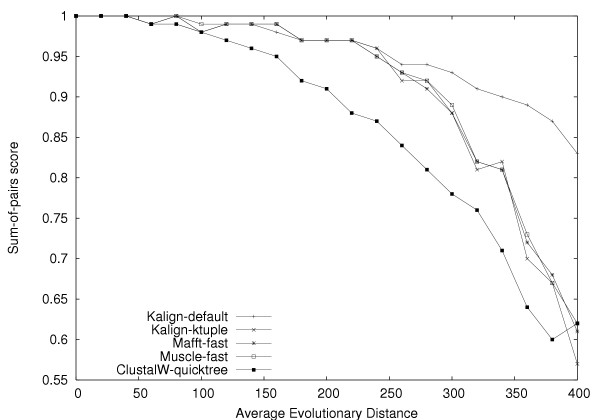
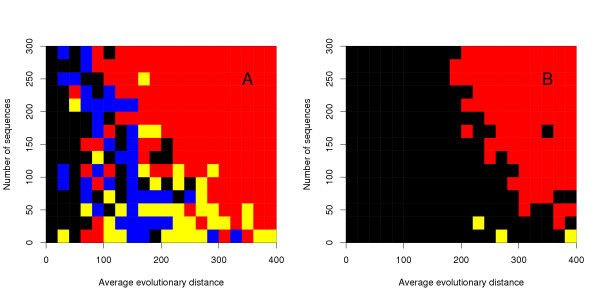
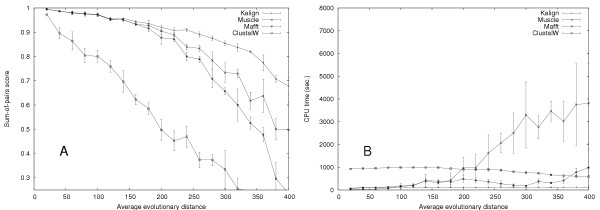
We developed Kalign, a method employing the Wu-Manber string-matching algorithm, to improve both the accuracy and speed of multiple sequence alignment. We compared the speed and accuracy of Kalign to other popular methods using Balibase, Prefab, and a new large test set. Kalign was as accurate as the best other methods on small alignments, but significantly more accurate when aligning large and distantly related sets of sequences. In our comparisons, Kalign was about 10 times faster than ClustalW and, depending on the alignment size, up to 50 times faster than popular iterative methods.
Kalign is a fast and robust alignment method. It is especially well suited for the increasingly important task of aligning large numbers of sequences.
多条蛋白质序列的比对是生物数据分析中的一个基本步骤。传统上,它被用于分析蛋白质家族的保守基序、系统发育、结构特性,以及提高同源性搜索的灵敏度。完整基因组序列的可得性增加了对多序列比对(MSA)程序的需求。当前的MSA方法存在要么不准确,要么计算成本过高,无法有效地应用于大规模比较基因组学的问题。
我们开发了Kalign,一种采用Wu-Manber字符串匹配算法的方法,以提高多序列比对的准确性和速度。我们使用Balibase、Prefab和一个新的大型测试集,将Kalign的速度和准确性与其他流行方法进行了比较。在小比对中,Kalign与其他最佳方法一样准确,但在比对大型和远缘相关的序列集时,准确性明显更高。在我们的比较中,Kalign比ClustalW快约10倍,并且根据比对大小,比流行的迭代方法快达50倍。
Kalign是一种快速且稳健的比对方法。它特别适合于比对大量序列这一日益重要的任务。