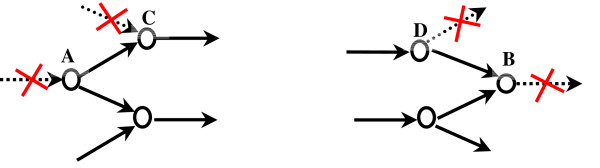Satish Kumar Vinay, Dasika Madhukar S, Maranas Costas D
Department of Industrial and Manufacturing Engineering, The Pennsylvania State University, University Park, PA 16802, USA.
BMC Bioinformatics. 2007 Jun 20;8:212. doi: 10.1186/1471-2105-8-212.
Currently, there exists tens of different microbial and eukaryotic metabolic reconstructions (e.g., Escherichia coli, Saccharomyces cerevisiae, Bacillus subtilis) with many more under development. All of these reconstructions are inherently incomplete with some functionalities missing due to the lack of experimental and/or homology information. A key challenge in the automated generation of genome-scale reconstructions is the elucidation of these gaps and the subsequent generation of hypotheses to bridge them.
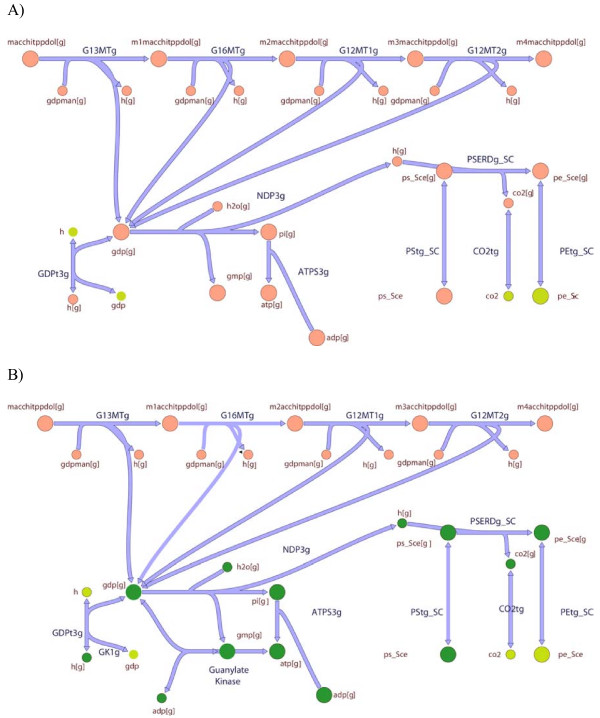
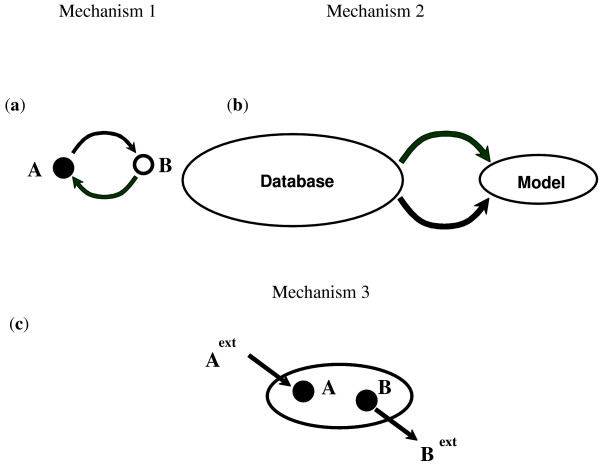
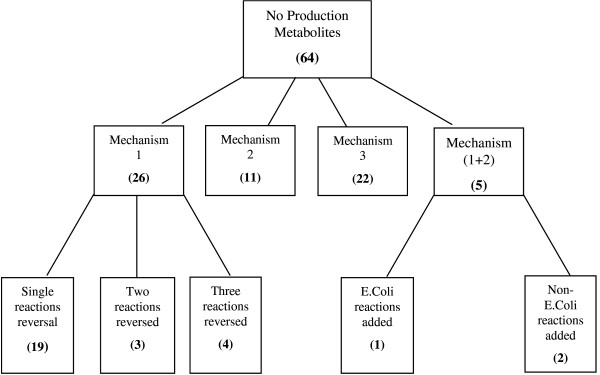
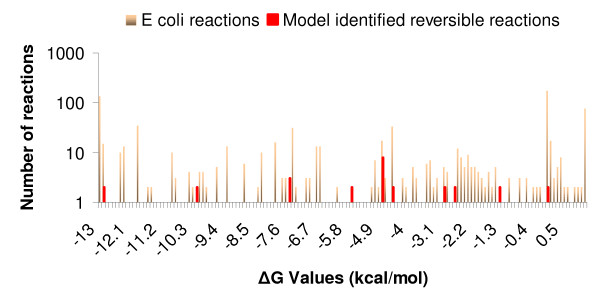
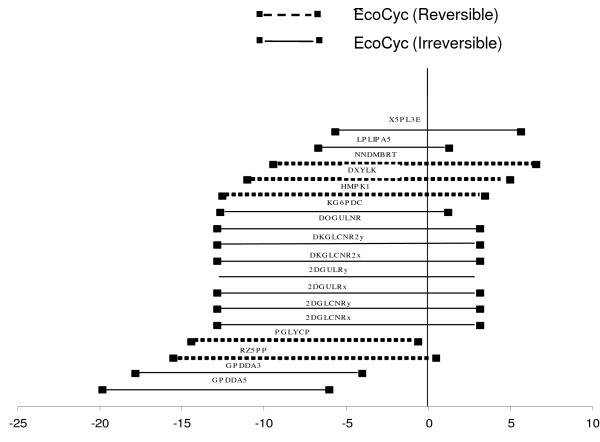
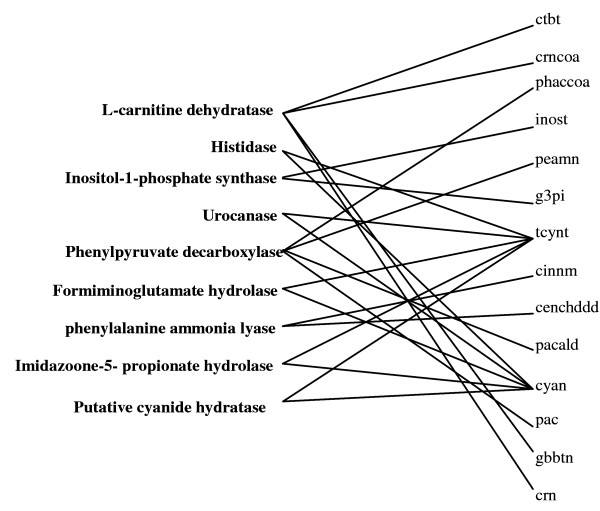
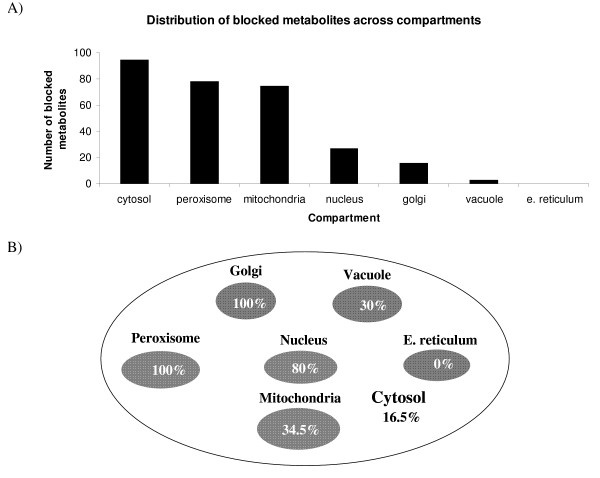
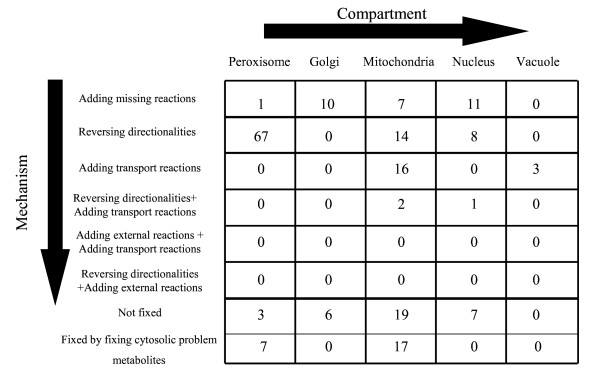
In this work, an optimization based procedure is proposed to identify and eliminate network gaps in these reconstructions. First we identify the metabolites in the metabolic network reconstruction which cannot be produced under any uptake conditions and subsequently we identify the reactions from a customized multi-organism database that restores the connectivity of these metabolites to the parent network using four mechanisms. This connectivity restoration is hypothesized to take place through four mechanisms: a) reversing the directionality of one or more reactions in the existing model, b) adding reaction from another organism to provide functionality absent in the existing model, c) adding external transport mechanisms to allow for importation of metabolites in the existing model and d) restore flow by adding intracellular transport reactions in multi-compartment models. We demonstrate this procedure for the genome- scale reconstruction of Escherichia coli and also Saccharomyces cerevisiae wherein compartmentalization of intra-cellular reactions results in a more complex topology of the metabolic network. We determine that about 10% of metabolites in E. coli and 30% of metabolites in S. cerevisiae cannot carry any flux. Interestingly, the dominant flow restoration mechanism is directionality reversals of existing reactions in the respective models.
We have proposed systematic methods to identify and fill gaps in genome-scale metabolic reconstructions. The identified gaps can be filled both by making modifications in the existing model and by adding missing reactions by reconciling multi-organism databases of reactions with existing genome-scale models. Computational results provide a list of hypotheses to be queried further and tested experimentally.
目前,存在数十种不同的微生物和真核生物代谢重建模型(例如大肠杆菌、酿酒酵母、枯草芽孢杆菌),还有更多模型正在开发中。由于缺乏实验和/或同源性信息,所有这些重建模型本质上都是不完整的,缺少一些功能。在自动生成基因组规模重建模型时,一个关键挑战是阐明这些差距,并随后生成假设来弥合它们。
在这项工作中,提出了一种基于优化的程序来识别和消除这些重建模型中的网络差距。首先,我们识别代谢网络重建中在任何摄取条件下都无法产生的代谢物,随后我们从一个定制的多生物体数据库中识别反应,该数据库使用四种机制恢复这些代谢物与母网络的连通性。这种连通性恢复被假设通过四种机制发生:a)反转现有模型中一个或多个反应的方向性,b)添加来自另一个生物体的反应以提供现有模型中不存在的功能,c)添加外部运输机制以允许在现有模型中输入代谢物,d)通过在多隔室模型中添加细胞内运输反应来恢复流量。我们展示了该程序在大肠杆菌和酿酒酵母的基因组规模重建中的应用,其中细胞内反应的区室化导致代谢网络的拓扑结构更加复杂。我们确定大肠杆菌中约10%的代谢物和酿酒酵母中30%的代谢物无法携带任何通量。有趣的是,主要的流量恢复机制是各个模型中现有反应的方向性反转。
我们提出了系统的方法来识别和填补基因组规模代谢重建中的差距。通过修改现有模型以及通过将多生物体反应数据库与现有基因组规模模型进行协调来添加缺失反应,可以填补所识别的差距。计算结果提供了一系列有待进一步查询和实验测试的假设。