Prakash Amol, Tompa Martin
Department of Computer Science and Engineering, University of Washington, Seattle, WA 98195-2350, USA.
Genome Biol. 2007;8(6):R124. doi: 10.1186/gb-2007-8-6-r124.
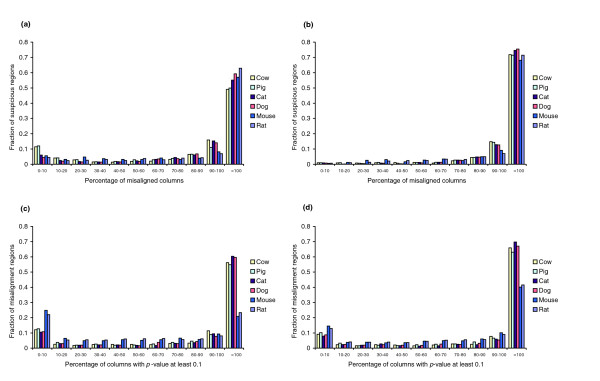
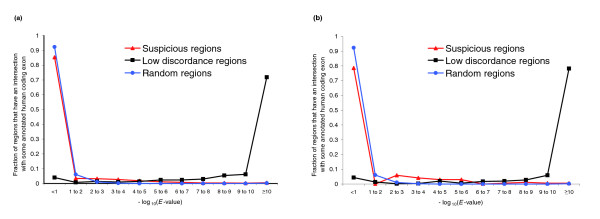
Whole-genome alignments are invaluable for comparative genomics. Before doing any comparative analysis on a region of interest, one must have confidence in that region's alignment. We provide a methodology to measure the accuracy of arbitrary regions of these alignments, and apply it to the UCSC Genome Browser's 17-vertebrate alignment. We identify 9.7% (21 Mbp) of the human chromosome 1 alignment as suspiciously aligned. We present independent evidence that many of these suspicious regions represent misalignments.
全基因组比对对于比较基因组学而言具有极高价值。在对感兴趣的区域进行任何比较分析之前,必须对该区域的比对结果有信心。我们提供了一种方法来衡量这些比对中任意区域的准确性,并将其应用于加州大学圣克鲁兹分校基因组浏览器的17种脊椎动物比对。我们确定人类1号染色体比对中有9.7%(21兆碱基对)的区域比对存在可疑之处。我们提供了独立证据表明这些可疑区域中有许多代表了错误比对。