Department of Computational Biology, Faculty of Biology, Adam Mickiewicz University in Poznan, Umultowska 89, 61-614, Poznan, Poland.
IDMEC, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001, Lisbon, Portugal.
Genome Biol. 2017 Oct 3;18(1):186. doi: 10.1186/s13059-017-1319-7.
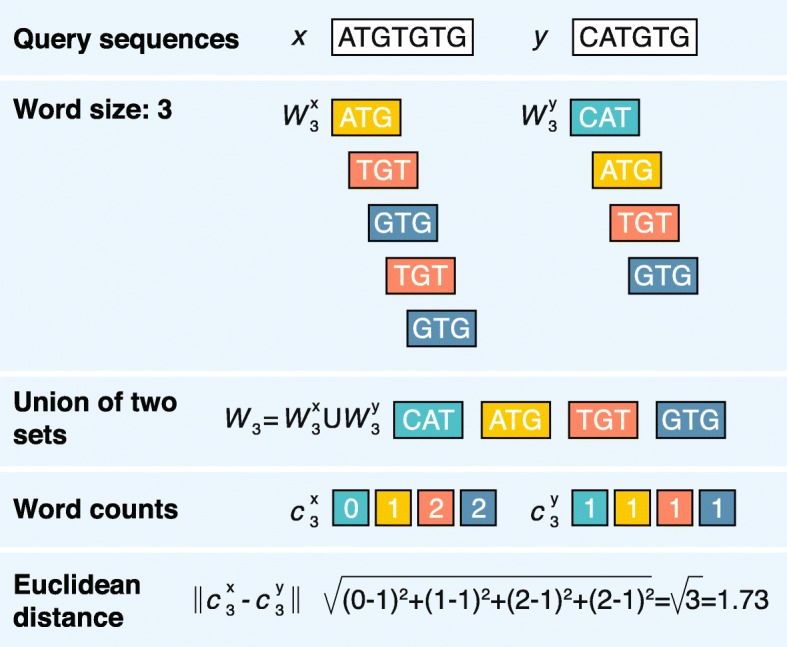
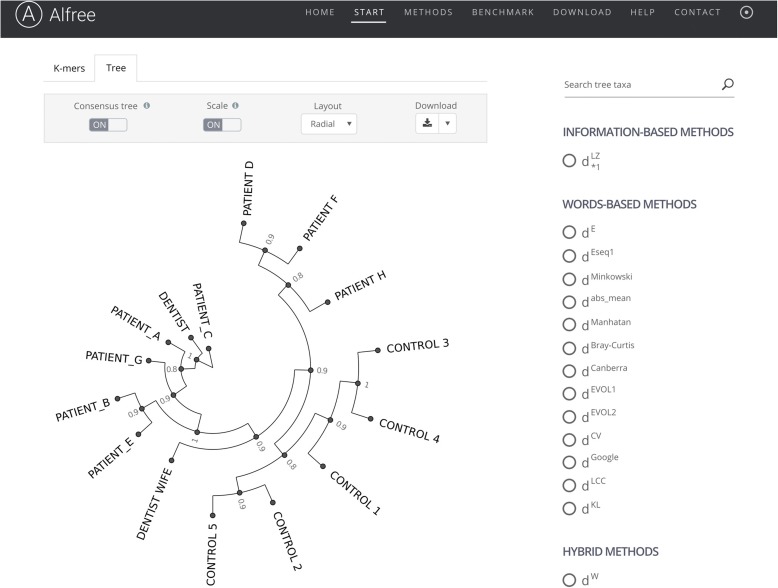
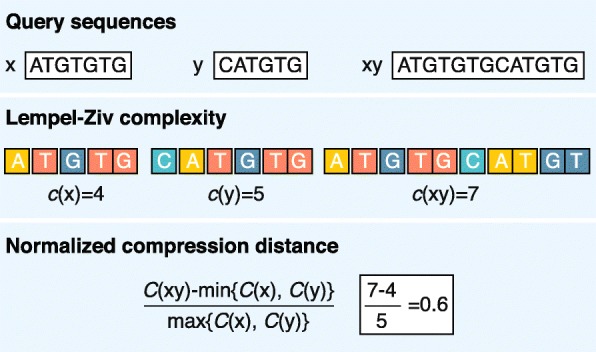
Alignment-free sequence analyses have been applied to problems ranging from whole-genome phylogeny to the classification of protein families, identification of horizontally transferred genes, and detection of recombined sequences. The strength of these methods makes them particularly useful for next-generation sequencing data processing and analysis. However, many researchers are unclear about how these methods work, how they compare to alignment-based methods, and what their potential is for use for their research. We address these questions and provide a guide to the currently available alignment-free sequence analysis tools.
无比对序列分析方法已经被应用于从全基因组系统发生到蛋白质家族分类、水平基因转移的鉴定以及重组序列的检测等各种问题。这些方法的优势使得它们特别适用于下一代测序数据的处理和分析。然而,许多研究人员并不清楚这些方法的工作原理、它们与基于比对的方法相比的优劣,以及它们在研究中的潜在用途。我们解决了这些问题,并提供了一份当前可用的无比对序列分析工具指南。