Tachmazidou Ioanna, Verzilli Claudio J, De Iorio Maria
Department of Epidemiology and Public Health, Imperial College London, United Kingdom.
PLoS Genet. 2007 Jul;3(7):e111. doi: 10.1371/journal.pgen.0030111.
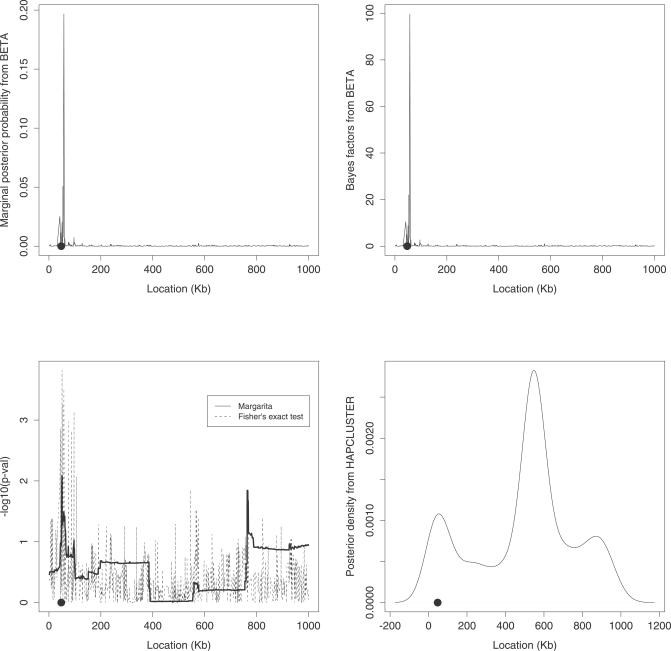
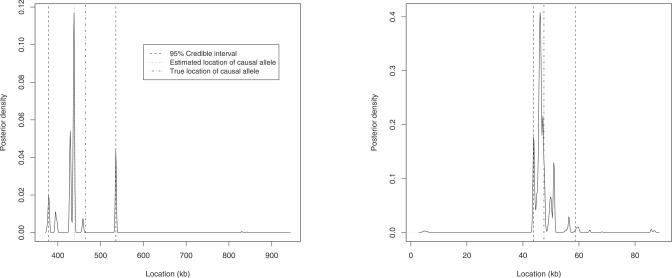
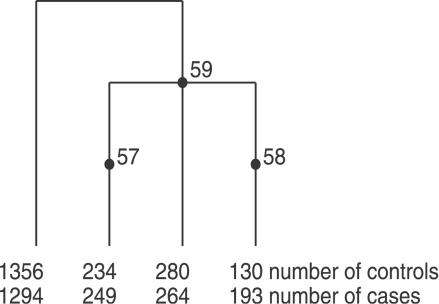
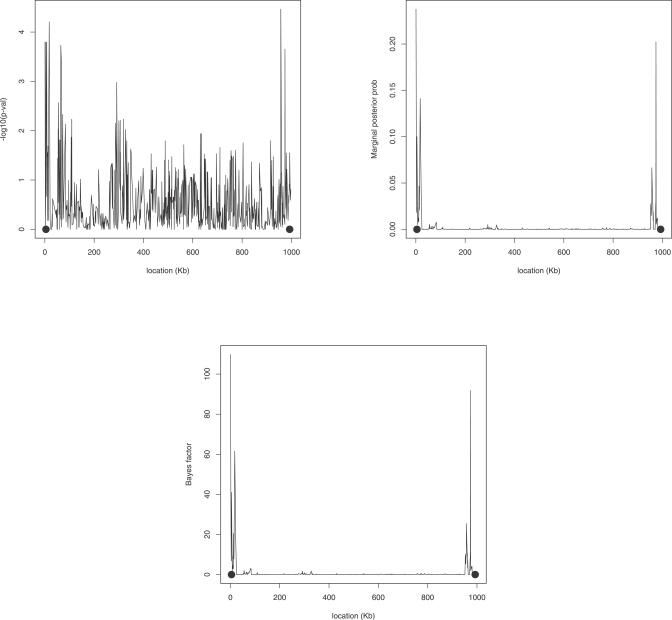
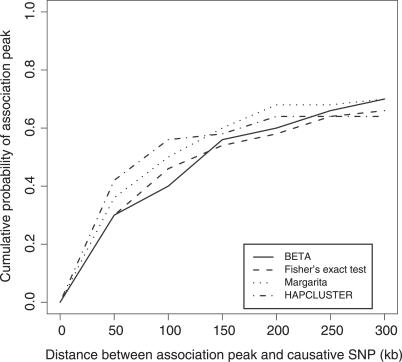
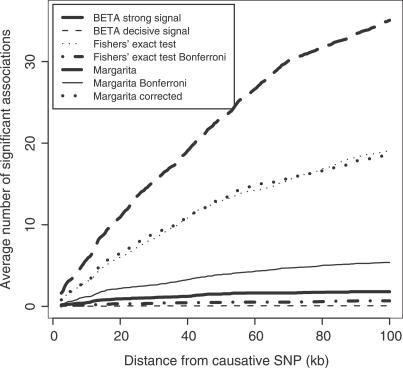
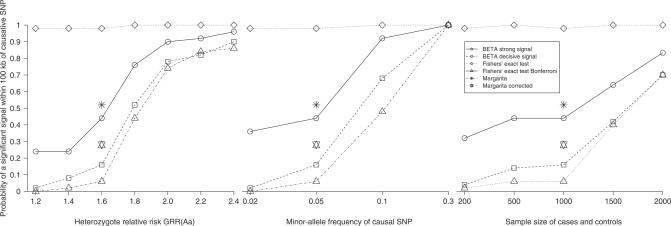
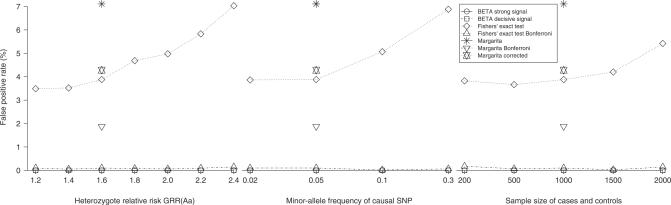
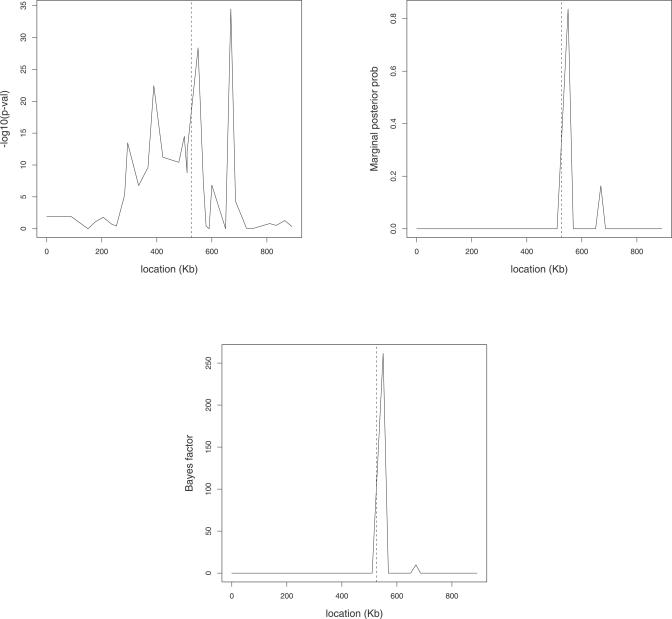
Multilocus analysis of single nucleotide polymorphism haplotypes is a promising approach to dissecting the genetic basis of complex diseases. We propose a coalescent-based model for association mapping that potentially increases the power to detect disease-susceptibility variants in genetic association studies. The approach uses Bayesian partition modelling to cluster haplotypes with similar disease risks by exploiting evolutionary information. We focus on candidate gene regions with densely spaced markers and model chromosomal segments in high linkage disequilibrium therein assuming a perfect phylogeny. To make this assumption more realistic, we split the chromosomal region of interest into sub-regions or windows of high linkage disequilibrium. The haplotype space is then partitioned into disjoint clusters, within which the phenotype-haplotype association is assumed to be the same. For example, in case-control studies, we expect chromosomal segments bearing the causal variant on a common ancestral background to be more frequent among cases than controls, giving rise to two separate haplotype clusters. The novelty of our approach arises from the fact that the distance used for clustering haplotypes has an evolutionary interpretation, as haplotypes are clustered according to the time to their most recent common ancestor. Our approach is fully Bayesian and we develop a Markov Chain Monte Carlo algorithm to sample efficiently over the space of possible partitions. We compare the proposed approach to both single-marker analyses and recently proposed multi-marker methods and show that the Bayesian partition modelling performs similarly in localizing the causal allele while yielding lower false-positive rates. Also, the method is computationally quicker than other multi-marker approaches. We present an application to real genotype data from the CYP2D6 gene region, which has a confirmed role in drug metabolism, where we succeed in mapping the location of the susceptibility variant within a small error.
单核苷酸多态性单倍型的多位点分析是剖析复杂疾病遗传基础的一种很有前景的方法。我们提出了一种基于合并的关联映射模型,该模型有可能提高在基因关联研究中检测疾病易感性变异的能力。该方法利用进化信息,通过贝叶斯分区建模对具有相似疾病风险的单倍型进行聚类。我们关注具有密集标记的候选基因区域,并假设存在完美系统发育,对其中处于高连锁不平衡状态的染色体片段进行建模。为了使这一假设更符合实际,我们将感兴趣的染色体区域划分为高连锁不平衡的子区域或窗口。然后将单倍型空间划分为不相交的聚类,在每个聚类中假设表型 - 单倍型关联是相同的。例如,在病例对照研究中,我们预计在共同祖先背景下携带因果变异的染色体片段在病例中比在对照中更常见,从而产生两个单独的单倍型聚类。我们方法的新颖之处在于,用于聚类单倍型的距离具有进化解释,因为单倍型是根据到其最近共同祖先的时间进行聚类的。我们的方法是完全贝叶斯的,并且我们开发了一种马尔可夫链蒙特卡罗算法,以便在可能的分区空间上进行高效采样。我们将所提出 的方法与单标记分析和最近提出的多标记方法进行了比较,结果表明贝叶斯分区建模在定位因果等位基因方面表现相似,同时产生较低的假阳性率。此外,该方法在计算上比其他多标记方法更快。我们展示了对来自CYP2D6基因区域的真实基因型数据的应用,该区域在药物代谢中具有确定的作用,我们成功地在小误差范围内定位了易感性变异的位置。