Kim Chang Sik
Bioinformatics Group, Turku Centre for Computer Science, Turku, Finland.
BMC Bioinformatics. 2007 Jul 13;8:251. doi: 10.1186/1471-2105-8-251.
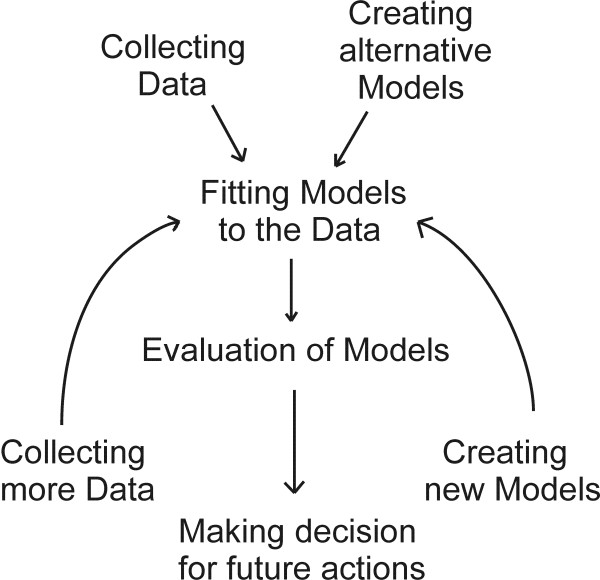
A reverse engineering of gene regulatory network with large number of genes and limited number of experimental data points is a computationally challenging task. In particular, reverse engineering using linear systems is an under-determined and ill conditioned problem, i.e. the amount of microarray data is limited and the solution is very sensitive to noise in the data. Therefore, the reverse engineering of gene regulatory networks with large number of genes and limited number of data points requires rigorous optimization algorithm.
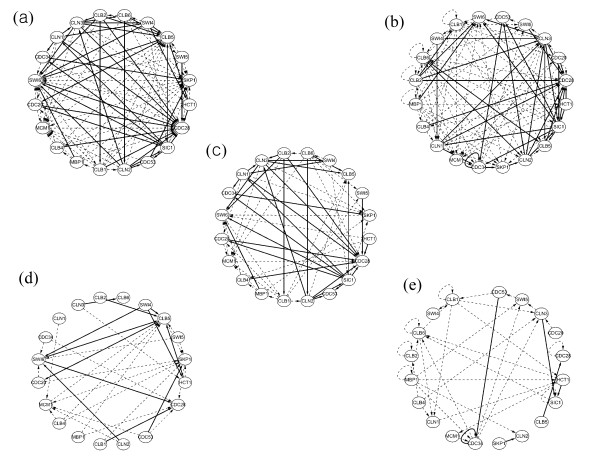
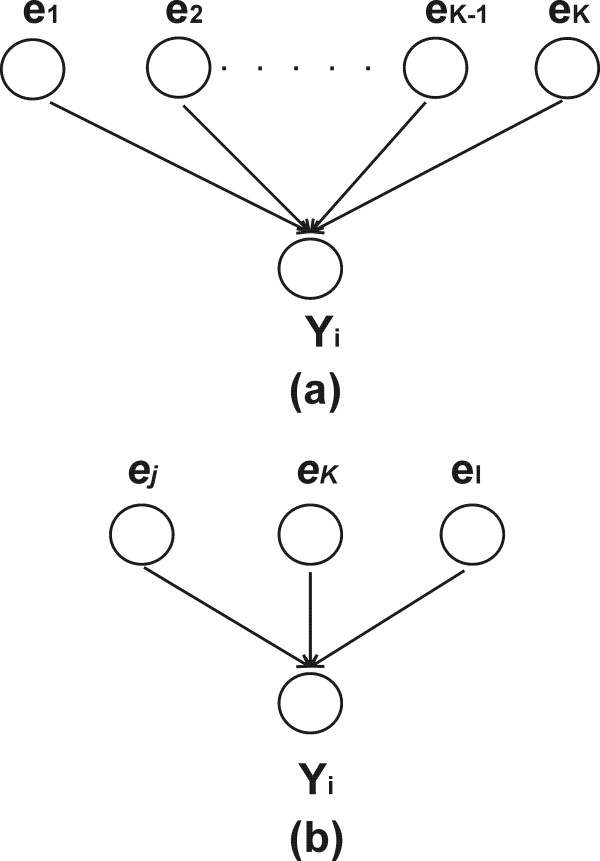
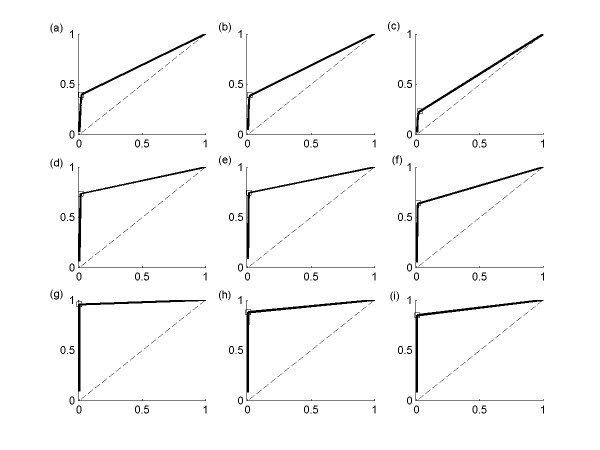
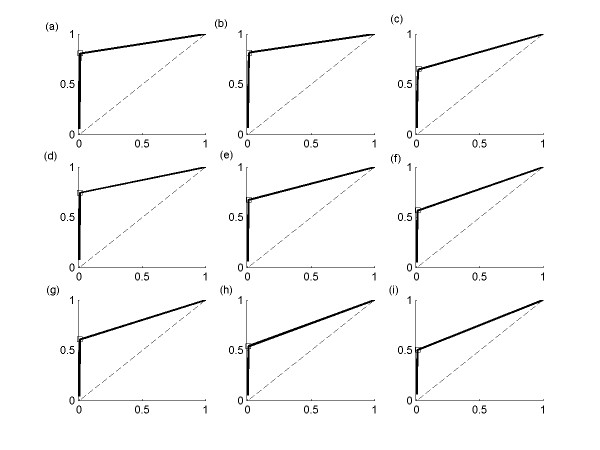
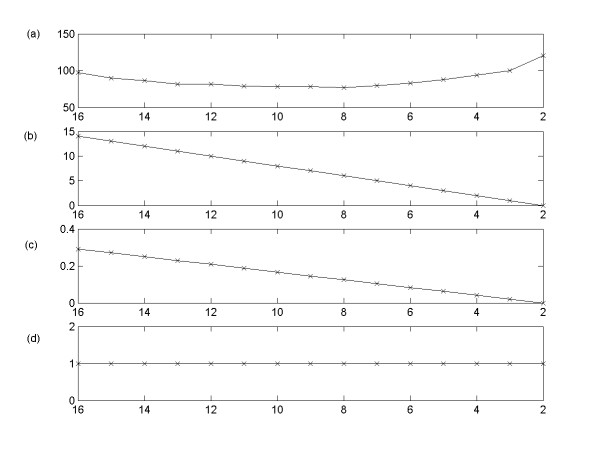
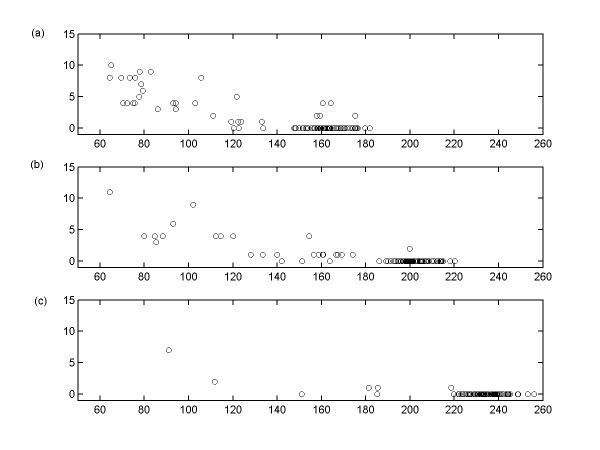
This study presents a novel algorithm for reverse engineering with linear systems. The proposed algorithm is a combination of the orthogonal least squares, second order derivative for network pruning, and Bayesian model comparison. In this study, the entire network is decomposed into a set of small networks that are defined as unit networks. The algorithm provides each unit network with P(D|Hi), which is used as confidence level. The unit network with higher P(D|Hi) has a higher confidence such that the unit network is correctly elucidated. Thus, the proposed algorithm is able to locate true positive interactions using P(D|Hi), which is a unique property of the proposed algorithm. The algorithm is evaluated with synthetic and Saccharomyces cerevisiae expression data using the dynamic Bayesian network. With synthetic data, it is shown that the performance of the algorithm depends on the number of genes, noise level, and the number of data points. With Yeast expression data, it is shown that there is remarkable number of known physical or genetic events among all interactions elucidated by the proposed algorithm. The performance of the algorithm is compared with Sparse Bayesian Learning algorithm using both synthetic and Saccharomyces cerevisiae expression data sets. The comparison experiments show that the algorithm produces sparser solutions with less false positives than Sparse Bayesian Learning algorithm.
From our evaluation experiments, we draw the conclusion as follows: 1) Simulation results show that the algorithm can be used to elucidate gene regulatory networks using limited number of experimental data points. 2) Simulation results also show that the algorithm is able to handle the problem with noisy data. 3) The experiment with Yeast expression data shows that the proposed algorithm reliably elucidates known physical or genetic events. 4) The comparison experiments show that the algorithm more efficiently performs than Sparse Bayesian Learning algorithm with noisy and limited number of data.
对具有大量基因和有限数量实验数据点的基因调控网络进行逆向工程是一项计算量极大的任务。特别是,使用线性系统进行逆向工程是一个欠定且病态的问题,即微阵列数据量有限,并且解对数据中的噪声非常敏感。因此,对具有大量基因和有限数量数据点的基因调控网络进行逆向工程需要严格的优化算法。
本研究提出了一种用于线性系统逆向工程的新算法。所提出的算法是正交最小二乘法、用于网络剪枝的二阶导数和贝叶斯模型比较的组合。在本研究中,整个网络被分解为一组定义为单元网络的小网络。该算法为每个单元网络提供P(D|Hi),其用作置信水平。具有较高P(D|Hi)的单元网络具有较高的置信度,从而该单元网络被正确阐明。因此,所提出的算法能够使用P(D|Hi)定位真正的正向相互作用,这是所提出算法的独特属性。使用动态贝叶斯网络对合成数据和酿酒酵母表达数据进行了算法评估。对于合成数据,结果表明算法的性能取决于基因数量、噪声水平和数据点数量。对于酵母表达数据,结果表明在所提出算法阐明的所有相互作用中存在大量已知的物理或遗传事件。使用合成数据和酿酒酵母表达数据集将该算法的性能与稀疏贝叶斯学习算法进行了比较。比较实验表明,与稀疏贝叶斯学习算法相比,该算法产生的解更稀疏,误报更少。
从我们的评估实验中,我们得出以下结论:1)模拟结果表明,该算法可用于使用有限数量的实验数据点阐明基因调控网络。2)模拟结果还表明,该算法能够处理有噪声数据的问题。3)酵母表达数据实验表明,所提出的算法能够可靠地阐明已知的物理或遗传事件。4)比较实验表明,在有噪声和有限数量数据的情况下,该算法比稀疏贝叶斯学习算法更高效。