Maia Rafaela M, Valente Valeria, Cunha Marco A V, Sousa Josane F, Araujo Daniela D, Silva Wilson A, Zago Marco A, Dias-Neto Emmanuel, Souza Sandro J, Simpson Andrew J G, Monesi Nadia, Ramos Ricardo G P, Espreafico Enilza M, Paçó-Larson Maria L
Departamento de Biologia Celular, Molecular e de Bioagentes Patogênicos, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brazil.
BMC Genomics. 2007 Jul 24;8:249. doi: 10.1186/1471-2164-8-249.
The sequencing of the D.melanogaster genome revealed an unexpected small number of genes (~ 14,000) indicating that mechanisms acting on generation of transcript diversity must have played a major role in the evolution of complex metazoans. Among the most extensively used mechanisms that accounts for this diversity is alternative splicing. It is estimated that over 40% of Drosophila protein-coding genes contain one or more alternative exons. A recent transcription map of the Drosophila embryogenesis indicates that 30% of the transcribed regions are unannotated, and that 1/3 of this is estimated as missed or alternative exons of previously characterized protein-coding genes. Therefore, the identification of the variety of expressed transcripts depends on experimental data for its final validation and is continuously being performed using different approaches. We applied the Open Reading Frame Expressed Sequence Tags (ORESTES) methodology, which is capable of generating cDNA data from the central portion of rare transcripts, in order to investigate the presence of hitherto unnanotated regions of Drosophila transcriptome.
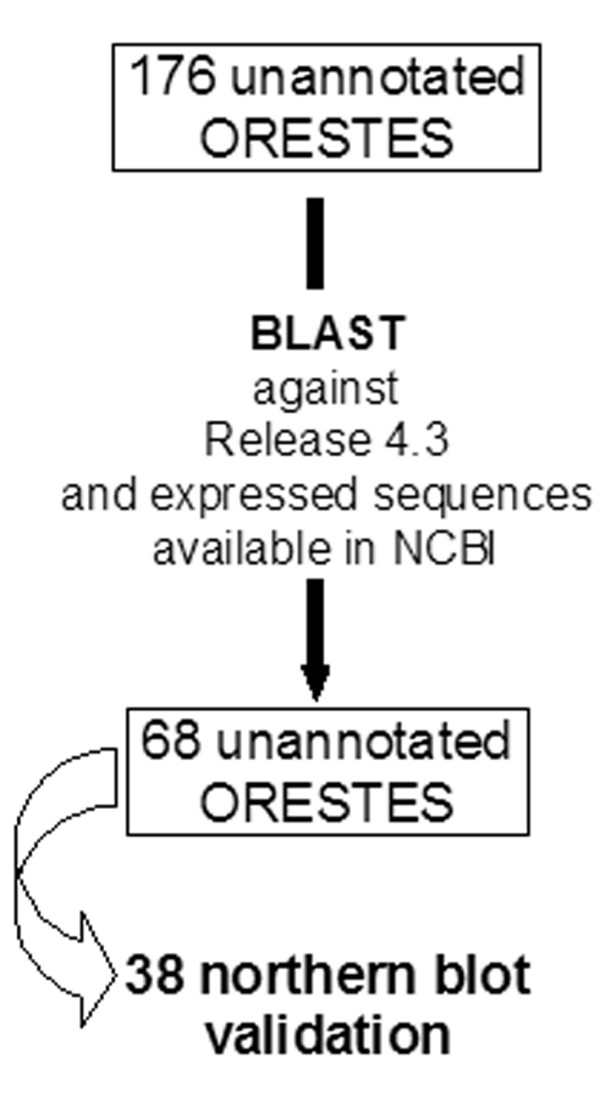
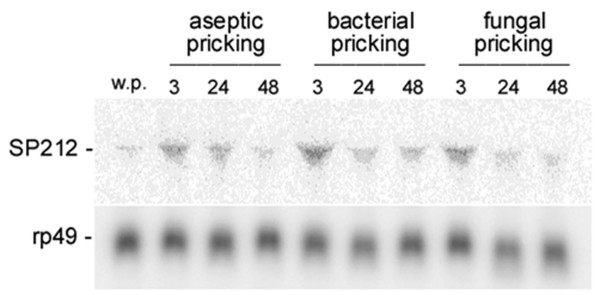
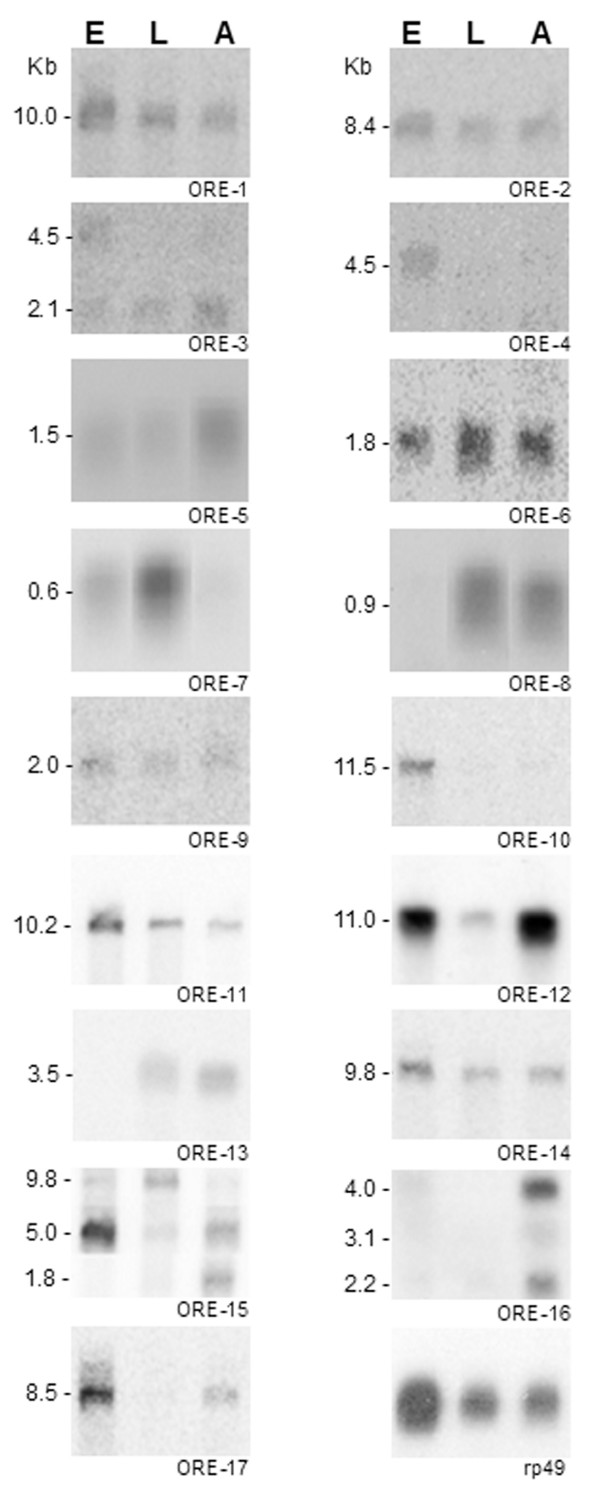
Bioinformatic analysis of 1,303 Drosophila ORESTES clusters identified 68 sequences derived from unannotated regions in the current Drosophila genome version (4.3). Of these, a set of 38 was analysed by polyA+ northern blot hybridization, validating 17 (50%) new exons of low abundance transcripts. For one of these ESTs, we obtained the cDNA encompassing the complete coding sequence of a new serine protease, named SP212. The SP212 gene is part of a serine protease gene cluster located in the chromosome region 88A12-B1. This cluster includes the predicted genes CG9631, CG9649 and CG31326, which were previously identified as up-regulated after immune challenges in genomic-scale microarray analysis. In agreement with the proposal that this locus is co-regulated in response to microorganisms infection, we show here that SP212 is also up-regulated upon injury.
Using the ORESTES methodology we identified 17 novel exons from low abundance Drosophila transcripts, and through a PCR approach the complete CDS of one of these transcripts was defined. Our results show that the computational identification and manual inspection are not sufficient to annotate a genome in the absence of experimentally derived data.
黑腹果蝇基因组测序显示基因数量出人意料地少(约14,000个),这表明作用于转录本多样性产生的机制在复杂后生动物的进化中一定发挥了主要作用。在导致这种多样性的最广泛使用的机制中,可变剪接是其中之一。据估计,超过40%的果蝇蛋白质编码基因包含一个或多个可变外显子。果蝇胚胎发育的最新转录图谱表明,30%的转录区域未被注释,其中1/3估计为先前已鉴定的蛋白质编码基因的遗漏或可变外显子。因此,各种表达转录本的鉴定依赖于实验数据进行最终验证,并且一直在使用不同方法不断进行。我们应用了开放阅读框表达序列标签(ORESTES)方法,该方法能够从稀有转录本的中央部分生成cDNA数据,以研究果蝇转录组中迄今未注释区域的存在情况。
对1303个果蝇ORESTES簇进行生物信息学分析,在当前果蝇基因组版本(4.3)中鉴定出68个源自未注释区域的序列。其中,一组38个通过polyA + Northern印迹杂交进行分析,验证了17个(50%)低丰度转录本的新外显子。对于其中一个EST,我们获得了包含一种新的丝氨酸蛋白酶完整编码序列的cDNA,该蛋白酶命名为SP212。SP212基因是位于染色体区域88A12 - B1的丝氨酸蛋白酶基因簇的一部分。该簇包括预测基因CG9631、CG9649和CG31326,这些基因在基因组规模微阵列分析中先前被鉴定为在免疫挑战后上调。与该基因座在响应微生物感染时共同调节的提议一致,我们在此表明SP212在受伤时也会上调。
使用ORESTES方法,我们从低丰度果蝇转录本中鉴定出17个新外显子,并通过PCR方法确定了其中一个转录本的完整CDS。我们的结果表明,在缺乏实验数据的情况下,计算鉴定和人工检查不足以注释基因组。