Su Emily Chia-Yu, Chiu Hua-Sheng, Lo Allan, Hwang Jenn-Kang, Sung Ting-Yi, Hsu Wen-Lian
Bioinformatics Program, Taiwan International Graduate Program, Academia Sinica, Taipei, Taiwan.
BMC Bioinformatics. 2007 Sep 8;8:330. doi: 10.1186/1471-2105-8-330.
Protein subcellular localization is crucial for genome annotation, protein function prediction, and drug discovery. Determination of subcellular localization using experimental approaches is time-consuming; thus, computational approaches become highly desirable. Extensive studies of localization prediction have led to the development of several methods including composition-based and homology-based methods. However, their performance might be significantly degraded if homologous sequences are not detected. Moreover, methods that integrate various features could suffer from the problem of low coverage in high-throughput proteomic analyses due to the lack of information to characterize unknown proteins.
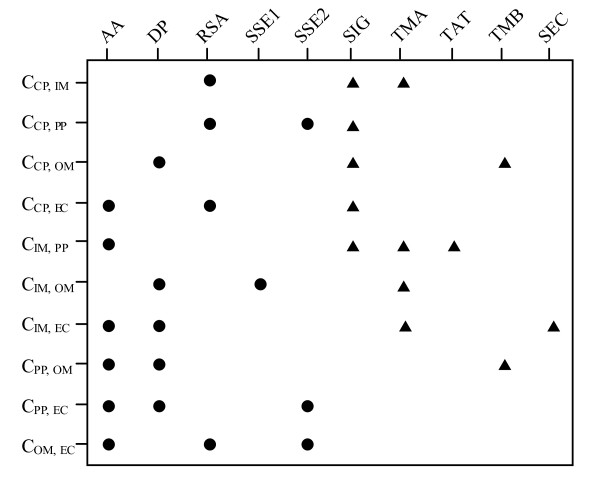
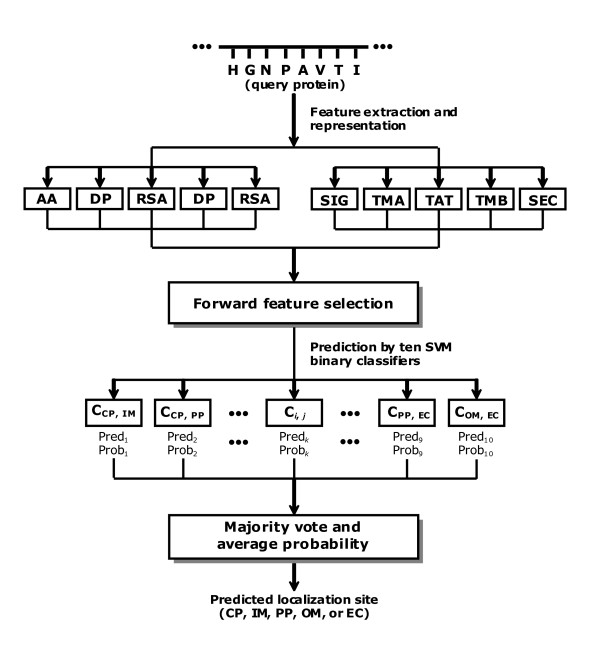
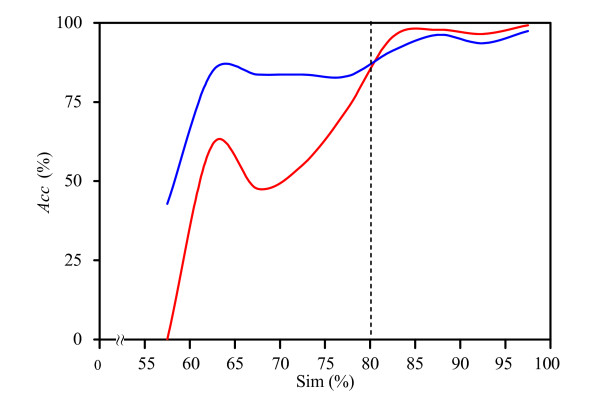
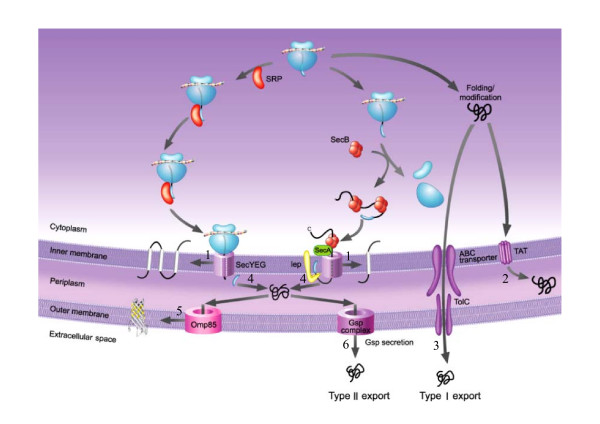
We propose a hybrid prediction method for Gram-negative bacteria that combines a one-versus-one support vector machines (SVM) model and a structural homology approach. The SVM model comprises a number of binary classifiers, in which biological features derived from Gram-negative bacteria translocation pathways are incorporated. In the structural homology approach, we employ secondary structure alignment for structural similarity comparison and assign the known localization of the top-ranked protein as the predicted localization of a query protein. The hybrid method achieves overall accuracy of 93.7% and 93.2% using ten-fold cross-validation on the benchmark data sets. In the assessment of the evaluation data sets, our method also attains accurate prediction accuracy of 84.0%, especially when testing on sequences with a low level of homology to the training data. A three-way data split procedure is also incorporated to prevent overestimation of the predictive performance. In addition, we show that the prediction accuracy should be approximately 85% for non-redundant data sets of sequence identity less than 30%.
Our results demonstrate that biological features derived from Gram-negative bacteria translocation pathways yield a significant improvement. The biological features are interpretable and can be applied in advanced analyses and experimental designs. Moreover, the overall accuracy of combining the structural homology approach is further improved, which suggests that structural conservation could be a useful indicator for inferring localization in addition to sequence homology. The proposed method can be used in large-scale analyses of proteomes.
蛋白质亚细胞定位对于基因组注释、蛋白质功能预测和药物发现至关重要。使用实验方法确定亚细胞定位耗时;因此,计算方法变得非常必要。对定位预测的广泛研究导致了几种方法的发展,包括基于组成和基于同源性的方法。然而,如果未检测到同源序列,它们的性能可能会显著下降。此外,由于缺乏表征未知蛋白质的信息,整合各种特征的方法在高通量蛋白质组分析中可能会面临覆盖率低的问题。
我们提出了一种针对革兰氏阴性菌的混合预测方法,该方法结合了一对一支持向量机(SVM)模型和结构同源性方法。SVM模型包含多个二元分类器,其中纳入了源自革兰氏阴性菌转运途径的生物学特征。在结构同源性方法中,我们采用二级结构比对进行结构相似性比较,并将排名最高的蛋白质的已知定位指定为查询蛋白质的预测定位。使用基准数据集进行十折交叉验证时,混合方法的总体准确率分别达到93.7%和93.2%。在评估数据集的评估中,我们的方法也达到了84.0%的准确预测准确率,特别是在对与训练数据同源性较低的序列进行测试时。还纳入了一种三分法数据分割程序以防止对预测性能的高估。此外,我们表明,对于序列同一性小于30%的非冗余数据集,预测准确率应约为85%。
我们的结果表明,源自革兰氏阴性菌转运途径的生物学特征有显著改进。这些生物学特征是可解释的,可应用于高级分析和实验设计。此外,结合结构同源性方法的总体准确率进一步提高,这表明除了序列同源性外,结构保守性可能是推断定位的有用指标。所提出的方法可用于蛋白质组的大规模分析。