Coolen Marcel W, Statham Aaron L, Gardiner-Garden Margaret, Clark Susan J
Cancer Program, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, Sydney 2010, New South Wales, Australia.
Nucleic Acids Res. 2007;35(18):e119. doi: 10.1093/nar/gkm662. Epub 2007 Sep 13.
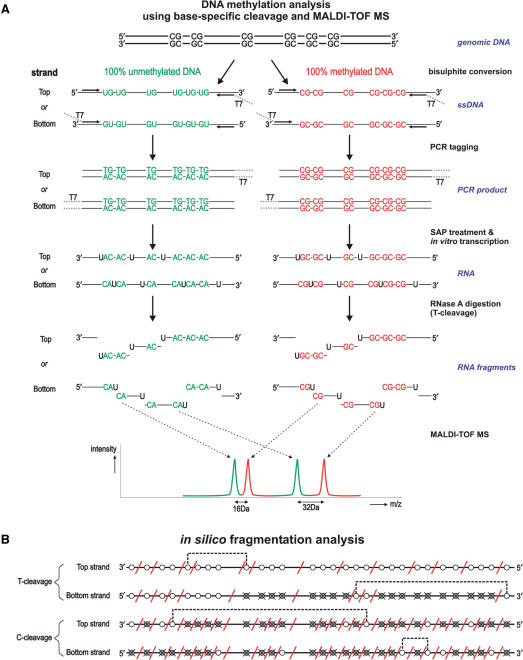
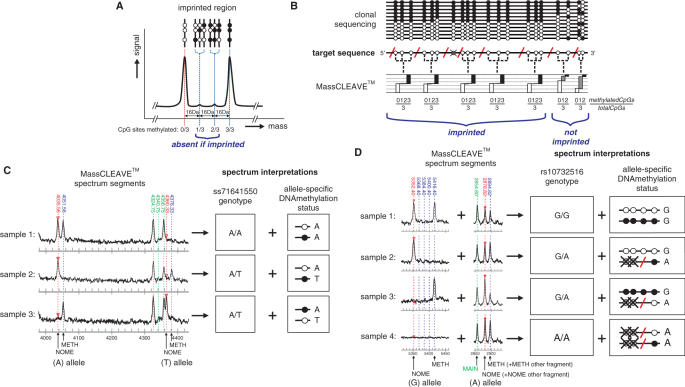
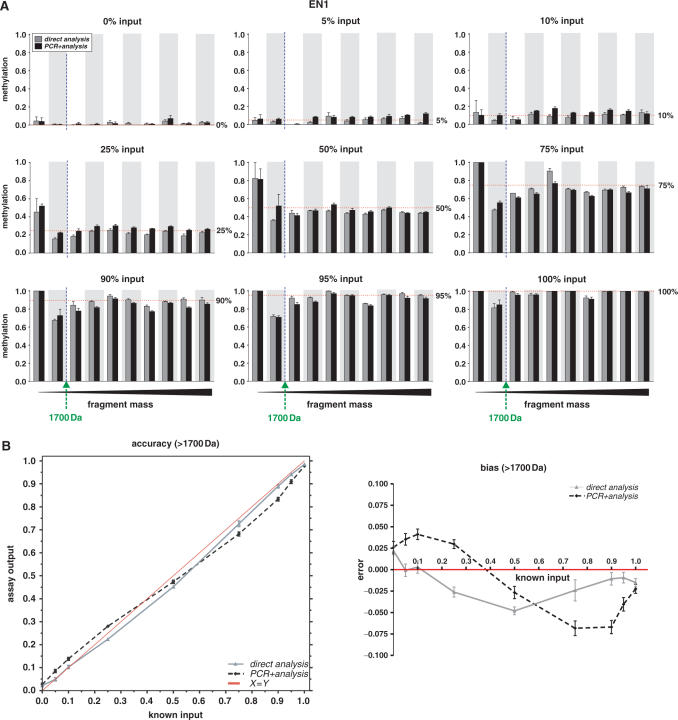
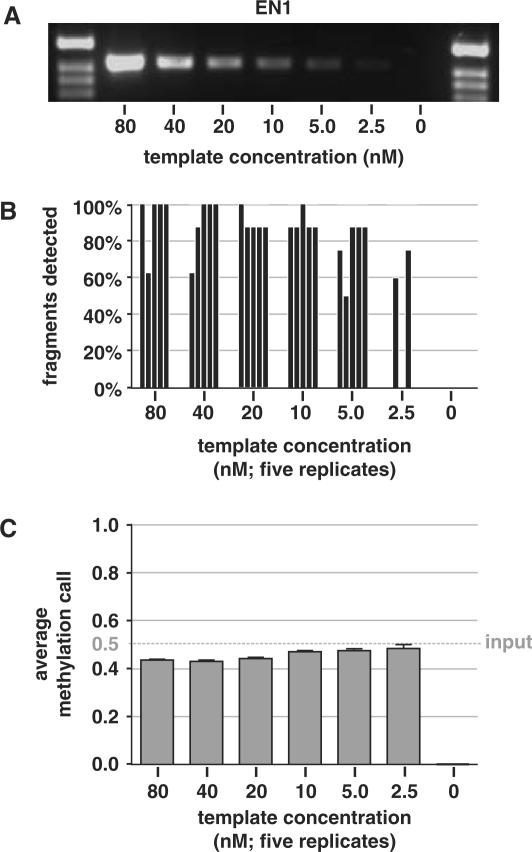
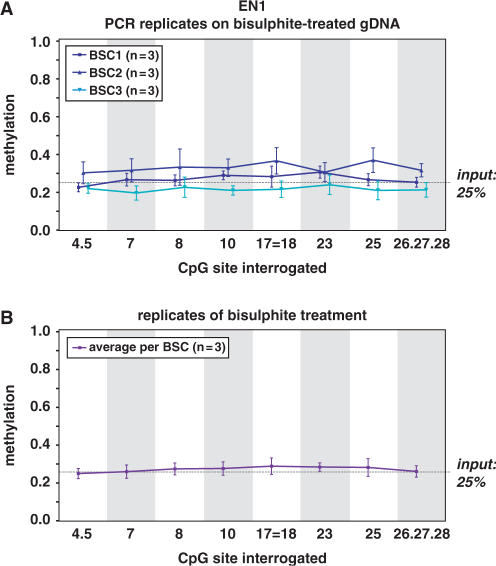
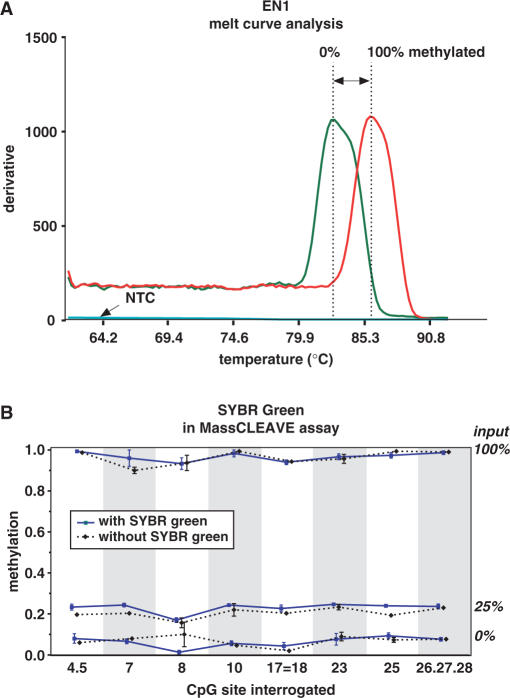
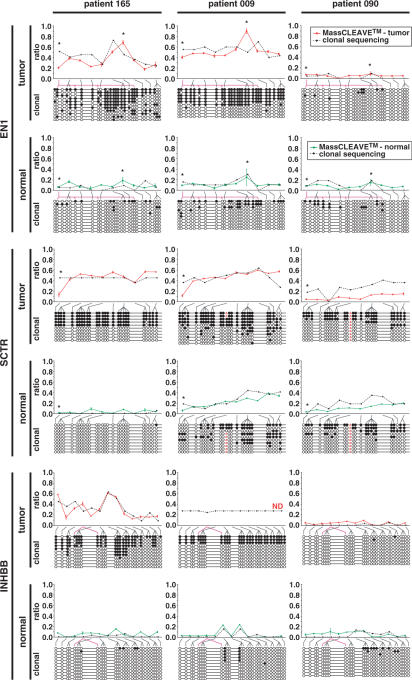
CpG methylation is a key component of the epigenome architecture that is associated with changes in gene expression without a change to the DNA sequence. Since the first reports on deregulation of DNA methylation, in diseases such as cancer, and the initiation of the Human Epigenome Project, an increasing need has arisen for a detailed, high-throughput and quantitative method of analysis to discover and validate normal and aberrant DNA methylation profiles in large sample cohorts. Here we present an improved protocol using base-specific fragmentation and MALDI-TOF mass spectrometry that enables a sensitive and high-throughput method of DNA methylation analysis, quantitative to 5% methylation for each informative CpG residue. We have determined the accuracy, variability and sensitivity of the protocol, implemented critical improvements in experimental design and interpretation of the data and developed a new formula to accurately measure CpG methylation. Key innovations now permit determination of differential and allele-specific methylation, such as in cancer and imprinting. The new protocol is ideally suitable for detailed DNA methylation analysis of multiple genomic regions and large sample cohorts that is critical for comprehensive profiling of normal and diseased human epigenomes.
CpG甲基化是表观基因组结构的关键组成部分,它与基因表达的变化相关,而DNA序列不变。自从首次报道DNA甲基化失调与癌症等疾病有关,以及人类表观基因组计划启动以来,对于一种详细、高通量且定量的分析方法的需求日益增加,以便在大量样本队列中发现和验证正常及异常的DNA甲基化谱。在此,我们展示了一种改进的方案,该方案使用碱基特异性片段化和基质辅助激光解吸电离飞行时间质谱(MALDI-TOF MS),实现了一种灵敏且高通量的DNA甲基化分析方法,对每个有信息的CpG残基的甲基化定量可达5%。我们已经确定了该方案的准确性、变异性和灵敏度,在实验设计和数据解释方面进行了关键改进,并开发了一个新公式来准确测量CpG甲基化。现在,关键创新允许确定差异甲基化和等位基因特异性甲基化,如在癌症和印记中。新方案非常适合对多个基因组区域和大量样本队列进行详细的DNA甲基化分析,这对于全面描绘正常和患病人类表观基因组至关重要。