Department of Life and Environmental Sciences, Chiba Institute of Technology, Narashino-shi, Chiba, Japan.
PLoS One. 2007 Sep 19;2(9):e905. doi: 10.1371/journal.pone.0000905.
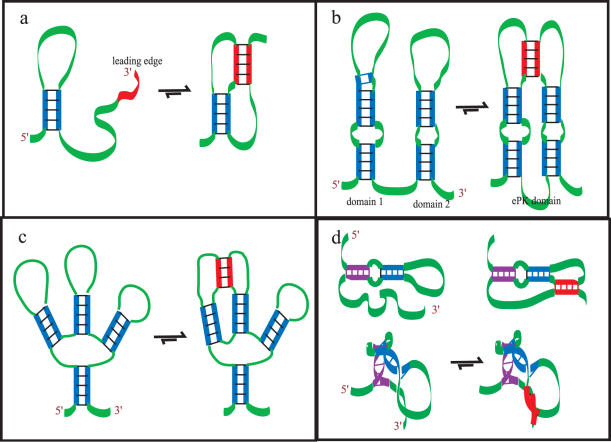
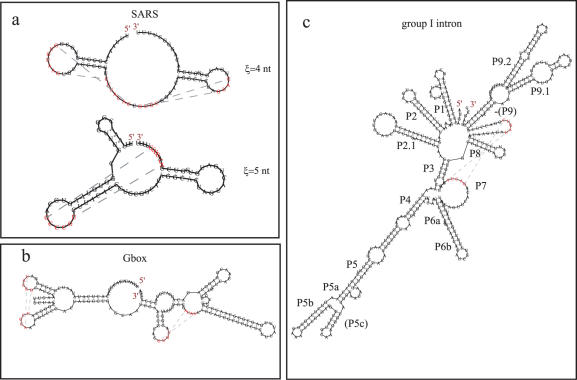
Predicting RNA secondary structure is often the first step to determining the structure of RNA. Prediction approaches have historically avoided searching for pseudoknots because of the extreme combinatorial and time complexity of the problem. Yet neglecting pseudoknots limits the utility of such approaches. Here, an algorithm utilizing structure mapping and thermodynamics is introduced for RNA pseudoknot prediction that finds the minimum free energy and identifies information about the flexibility of the RNA. The heuristic approach takes advantage of the 5' to 3' folding direction of many biological RNA molecules and is consistent with the hierarchical folding hypothesis and the contact order model. Mapping methods are used to build and analyze the folded structure for pseudoknots and to add important 3D structural considerations. The program can predict some well known pseudoknot structures correctly. The results of this study suggest that many functional RNA sequences are optimized for proper folding. They also suggest directions we can proceed in the future to achieve even better results.
预测 RNA 二级结构通常是确定 RNA 结构的第一步。由于问题的组合和时间复杂度极高,预测方法在历史上一直避免搜索假结。然而,忽略假结限制了此类方法的实用性。本文介绍了一种利用结构映射和热力学的 RNA 假结预测算法,该算法可找到最小自由能并识别 RNA 灵活性的信息。启发式方法利用了许多生物 RNA 分子的 5' 到 3' 折叠方向,与层次折叠假说和接触顺序模型一致。映射方法用于构建和分析假结的折叠结构,并添加重要的 3D 结构考虑因素。该程序可以正确预测一些已知的假结结构。这项研究的结果表明,许多功能 RNA 序列经过优化以实现正确折叠。它们还表明了我们未来可以继续努力以获得更好结果的方向。