Didier Gilles, Guziolowski Carito
Institut de Mathématiques de Luminy, 163 avenue de Luminy, Case 907, 13288 Marseille Cedex 9, France.
Algorithms Mol Biol. 2007 Sep 19;2:11. doi: 10.1186/1748-7188-2-11.
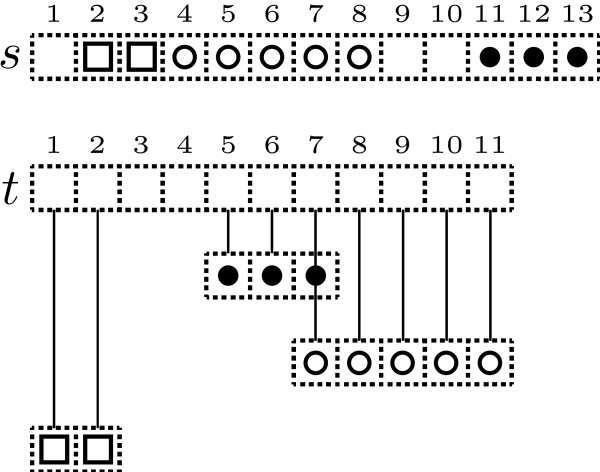
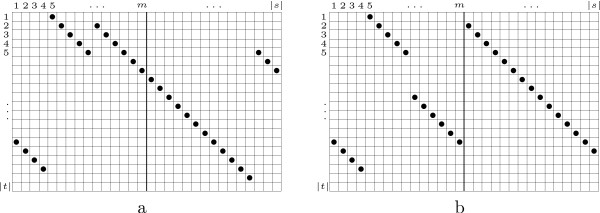
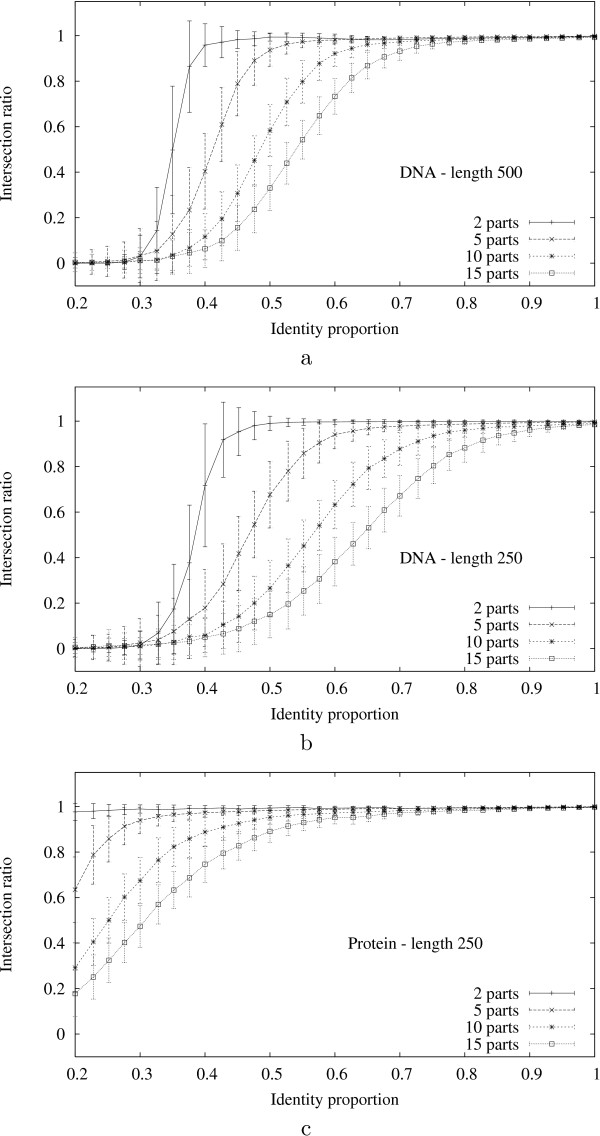
We present the N-map method, a pairwise and asymmetrical approach which allows us to compare sequences by taking into account evolutionary events that produce shuffled, reversed or repeated elements. Basically, the optimal N-map of a sequence s over a sequence t is the best way of partitioning the first sequence into N parts and placing them, possibly complementary reversed, over the second sequence in order to maximize the sum of their gapless alignment scores.
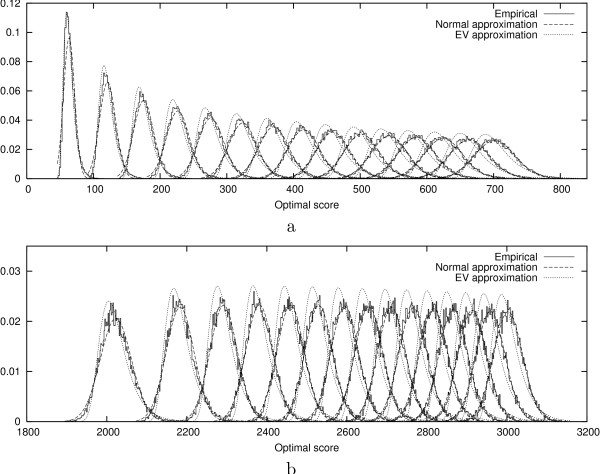
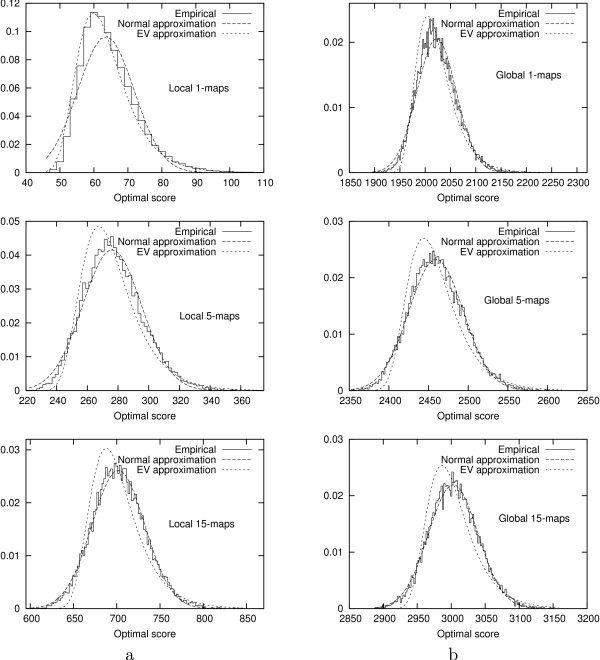
We introduce an algorithm computing an optimal N-map with time complexity O (|s| x |t| x N) using O (|s| x |t| x N) memory space. Among all the numbers of parts taken in a reasonable range, we select the value N for which the optimal N-map has the most significant score. To evaluate this significance, we study the empirical distributions of the scores of optimal N-maps and show that they can be approximated by normal distributions with a reasonable accuracy. We test the functionality of the approach over random sequences on which we apply artificial evolutionary events.
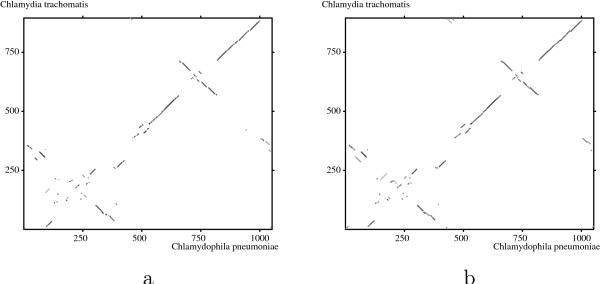
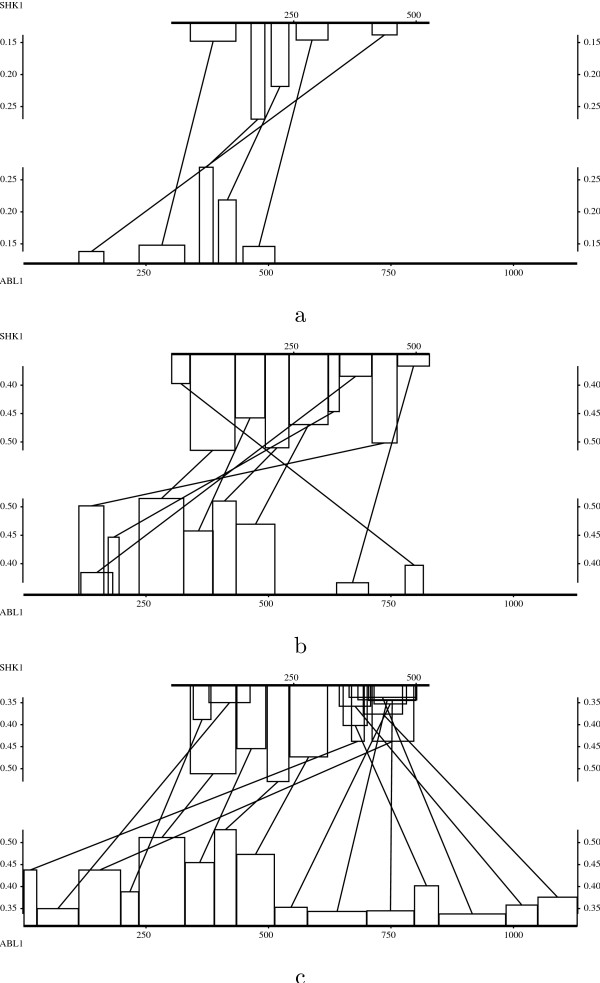
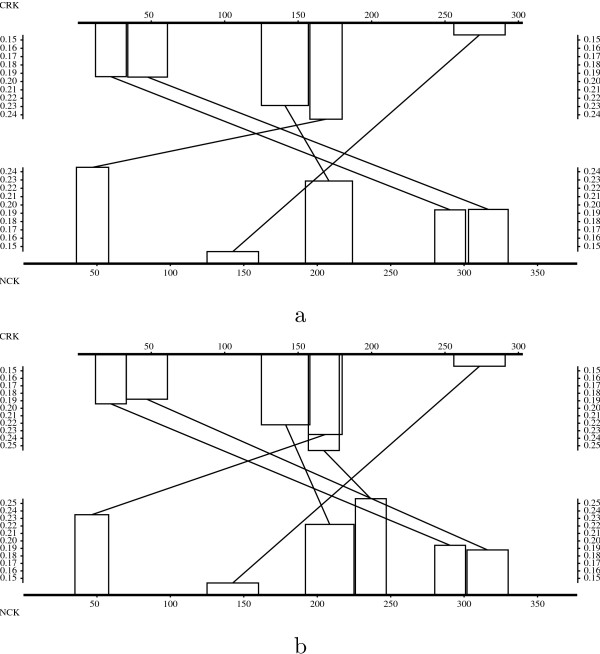
The method is illustrated with four case studies of pairs of sequences involving non-standard evolutionary events.
我们提出了N映射方法,这是一种成对且不对称的方法,通过考虑产生洗牌、反转或重复元素的进化事件,使我们能够比较序列。基本上,序列s相对于序列t的最优N映射是将第一个序列划分为N个部分并将它们(可能是互补反转的)放置在第二个序列上的最佳方式,以便最大化它们的无间隙比对分数之和。
我们引入了一种算法,该算法使用O(|s|×|t|×N)的内存空间,以O(|s|×|t|×N)的时间复杂度计算最优N映射。在合理范围内选取的所有部分数量中,我们选择最优N映射得分最高的N值。为了评估这种显著性,我们研究了最优N映射得分的经验分布,并表明它们可以以合理的精度用正态分布近似。我们在应用了人工进化事件的随机序列上测试了该方法的功能。
该方法通过涉及非标准进化事件的四对序列的案例研究进行了说明。