Fan Xiaodan, Zhu Jun, Schadt Eric E, Liu Jun S
Department of Statistics, Harvard University, Boston, MA, USA.
BMC Bioinformatics. 2007 Oct 5;8:374. doi: 10.1186/1471-2105-8-374.
An important goal of comparative genomics is the identification of functional elements through conservation analysis. Phylo-HMM was recently introduced to detect conserved elements based on multiple genome alignments, but the method has not been rigorously evaluated.
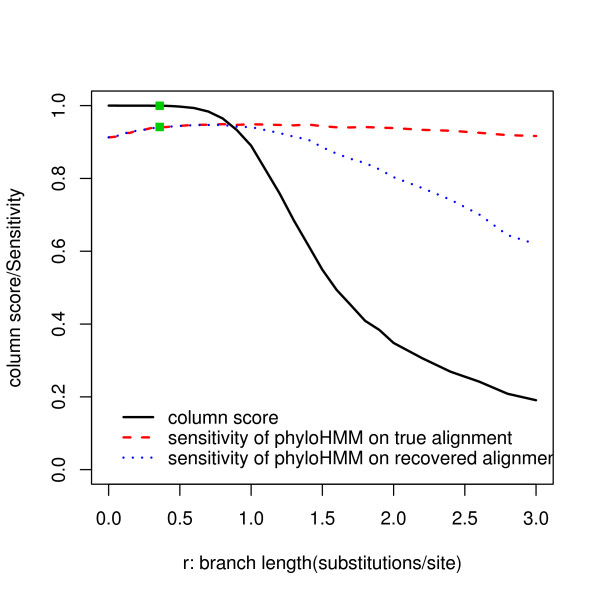
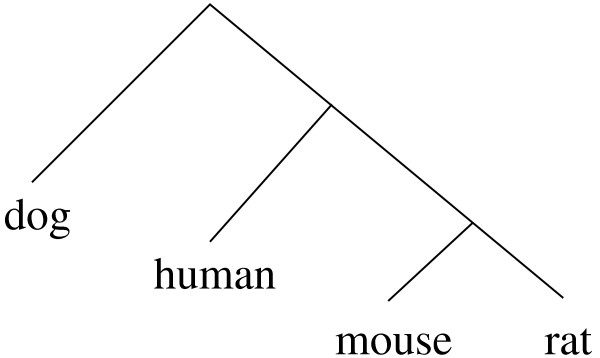
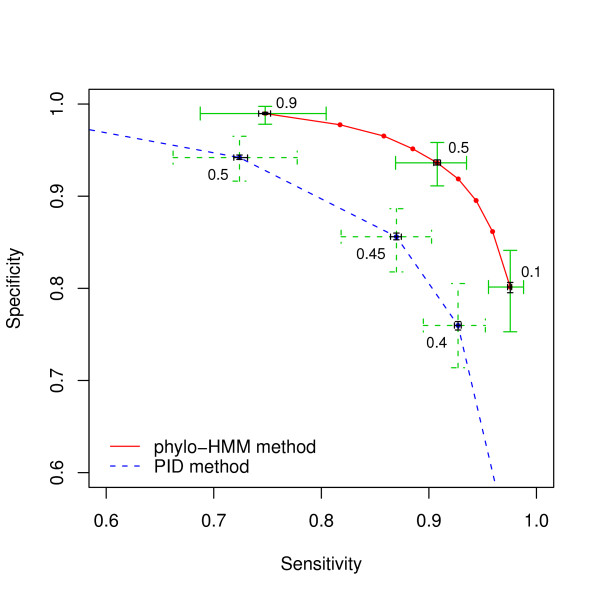
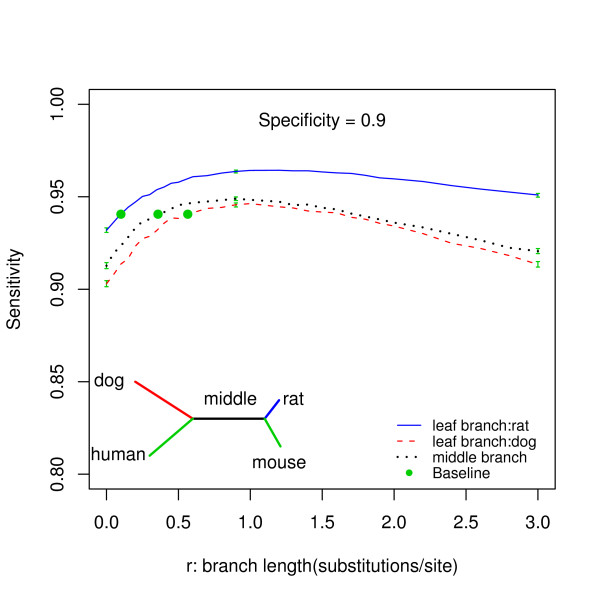
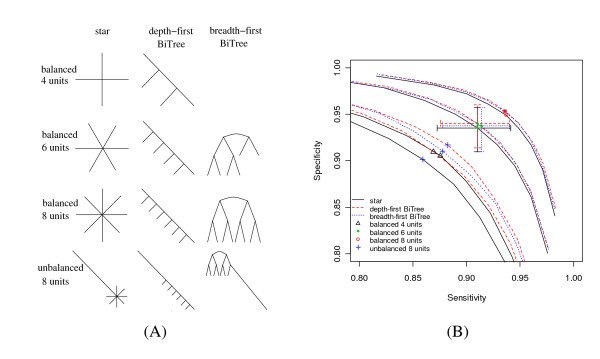
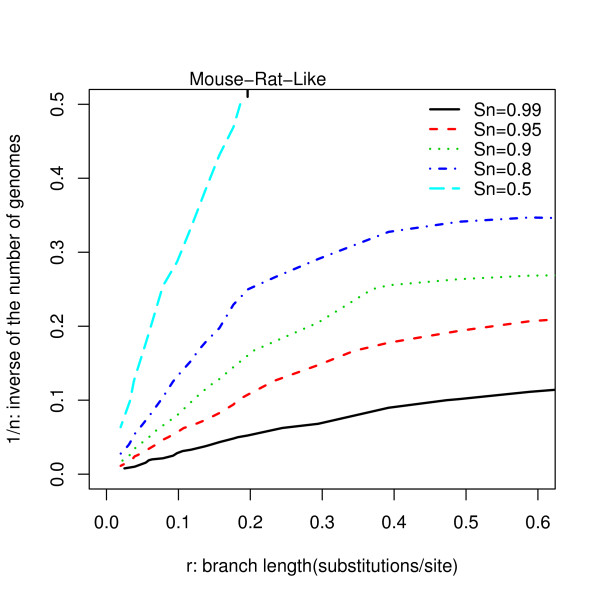
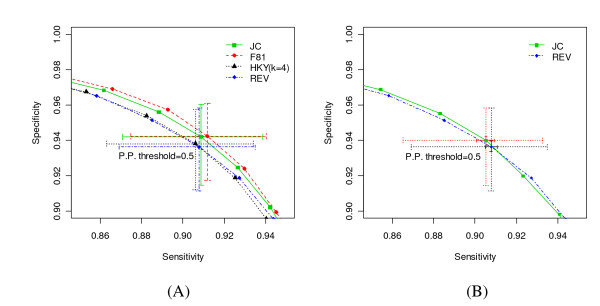
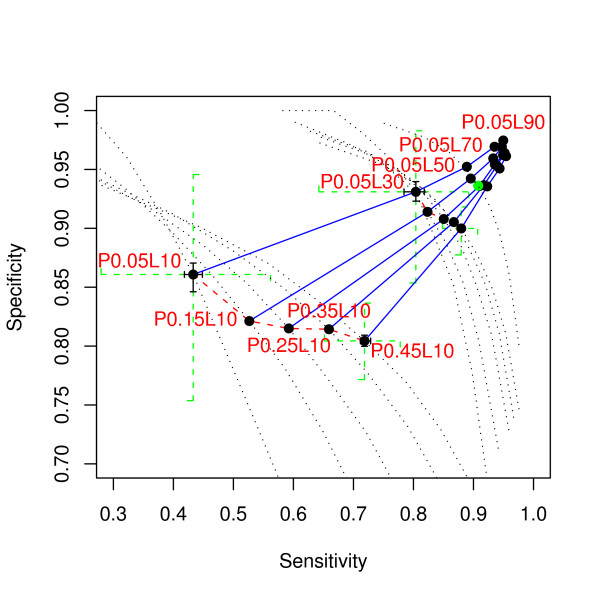
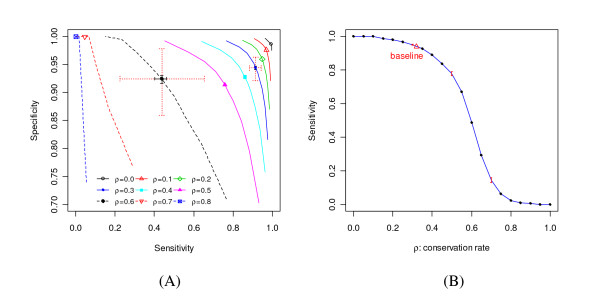
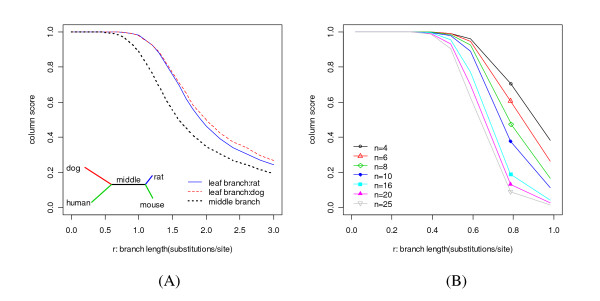
We report here a simulation study to investigate the power of phylo-HMM. We show that the power of the phylo-HMM approach depends on many factors, the most important being the number of species-specific genomes used and evolutionary distances between pairs of species. This finding is consistent with results reported by other groups for simpler comparative genomics models. In addition, the conservation ratio of conserved elements and the expected length of the conserved elements are also major factors. In contrast, the influence of the topology and the nucleotide substitution model are relatively minor factors.
Our results provide for general guidelines on how to select the number of genomes and their evolutionary distance in comparative genomics studies, as well as the level of power we can expect under different parameter settings.
比较基因组学的一个重要目标是通过保守性分析来识别功能元件。系统发育隐马尔可夫模型(Phylo-HMM)最近被引入用于基于多个基因组比对来检测保守元件,但该方法尚未得到严格评估。
我们在此报告一项模拟研究,以探究Phylo-HMM的效能。我们表明,Phylo-HMM方法的效能取决于许多因素,其中最重要的是所使用的物种特异性基因组的数量以及物种对之间的进化距离。这一发现与其他研究小组针对更简单的比较基因组学模型所报告的结果一致。此外,保守元件的保守率和保守元件的预期长度也是主要因素。相比之下,拓扑结构和核苷酸替换模型的影响相对较小。
我们的结果为在比较基因组学研究中如何选择基因组数量及其进化距离提供了一般指导原则,以及在不同参数设置下我们可以预期的效能水平。