Marioni John C, Thorne Natalie P, Valsesia Armand, Fitzgerald Tomas, Redon Richard, Fiegler Heike, Andrews T Daniel, Stranger Barbara E, Lynch Andrew G, Dermitzakis Emmanouil T, Carter Nigel P, Tavaré Simon, Hurles Matthew E
Computational Biology Group, Department of Applied Mathematics and Theoretical Physics, University of Cambridge, Centre for Mathematical Sciences, Wilberforce Road, Cambridge CB3 0WA, UK.
Genome Biol. 2007;8(10):R228. doi: 10.1186/gb-2007-8-10-r228.
Large-scale high throughput studies using microarray technology have established that copy number variation (CNV) throughout the genome is more frequent than previously thought. Such variation is known to play an important role in the presence and development of phenotypes such as HIV-1 infection and Alzheimer's disease. However, methods for analyzing the complex data produced and identifying regions of CNV are still being refined.
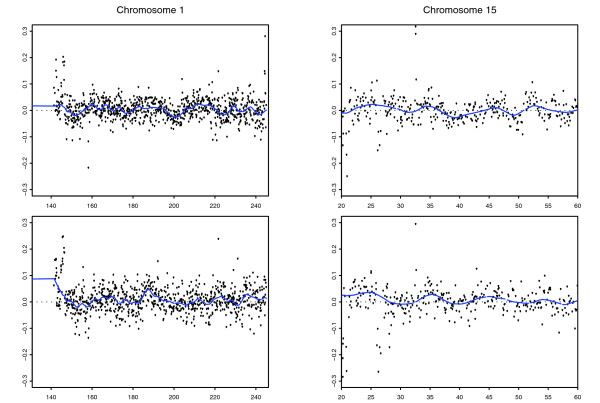
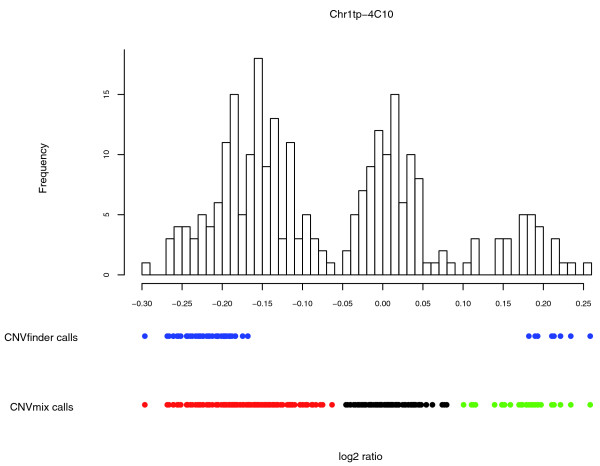
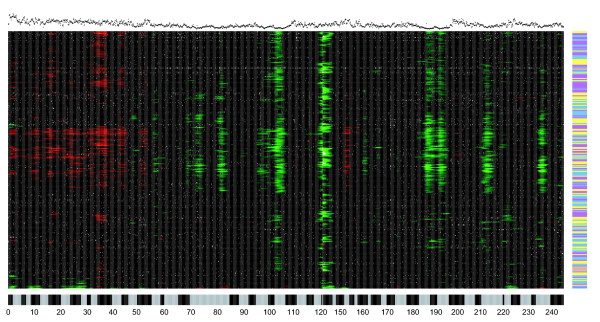
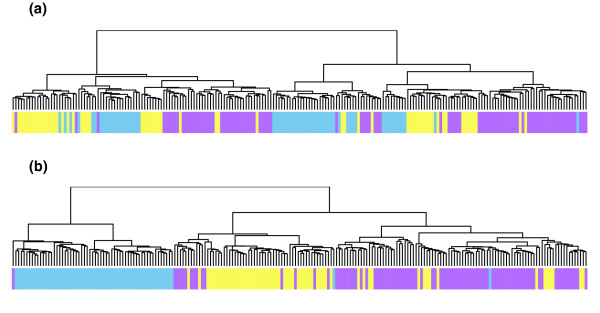
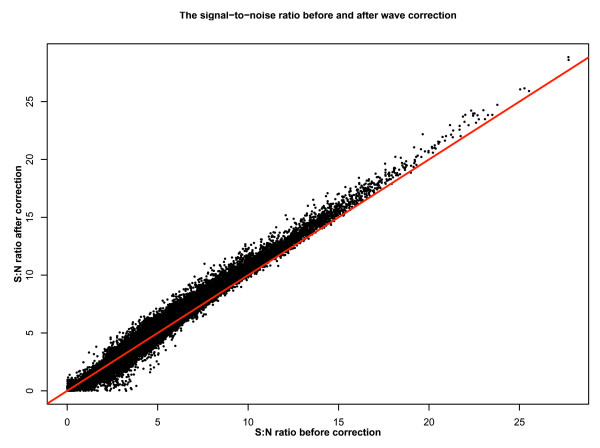
We describe the presence of a genome-wide technical artifact, spatial autocorrelation or 'wave', which occurs in a large dataset used to determine the location of CNV across the genome. By removing this artifact we are able to obtain both a more biologically meaningful clustering of the data and an increase in the number of CNVs identified by current calling methods without a major increase in the number of false positives detected. Moreover, removing this artifact is critical for the development of a novel model-based CNV calling algorithm - CNVmix - that uses cross-sample information to identify regions of the genome where CNVs occur. For regions of CNV that are identified by both CNVmix and current methods, we demonstrate that CNVmix is better able to categorize samples into groups that represent copy number gains or losses.
Removing artifactual 'waves' (which appear to be a general feature of array comparative genomic hybridization (aCGH) datasets) and using cross-sample information when identifying CNVs enables more biological information to be extracted from aCGH experiments designed to investigate copy number variation in normal individuals.
使用微阵列技术的大规模高通量研究已证实,全基因组范围内的拷贝数变异(CNV)比之前认为的更为常见。已知这种变异在诸如HIV-1感染和阿尔茨海默病等表型的出现和发展中起着重要作用。然而,用于分析所产生的复杂数据以及识别CNV区域的方法仍在不断完善。
我们描述了一种全基因组范围的技术假象,即空间自相关或“波”,它出现在用于确定全基因组CNV位置的一个大型数据集中。通过去除这种假象,我们既能获得更具生物学意义的数据聚类,又能在当前的检测方法中识别出更多的CNV,同时检测到的假阳性数量没有大幅增加。此外,去除这种假象对于开发一种基于模型的新型CNV检测算法——CNVmix至关重要,该算法利用跨样本信息来识别基因组中发生CNV的区域。对于通过CNVmix和当前方法都识别出的CNV区域,我们证明CNVmix能更好地将样本分类为代表拷贝数增加或减少的组。
去除人为的“波”(这似乎是阵列比较基因组杂交(aCGH)数据集的一个普遍特征),并在识别CNV时使用跨样本信息,能够从旨在研究正常个体拷贝数变异的aCGH实验中提取更多生物学信息。