Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA.
Department of Oncology The Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
BMC Cancer. 2020 Sep 7;20(1):856. doi: 10.1186/s12885-020-07304-3.
Germline copy number variants (CNVs) increase risk for many diseases, yet detection of CNVs and quantifying their contribution to disease risk in large-scale studies is challenging due to biological and technical sources of heterogeneity that vary across the genome within and between samples.
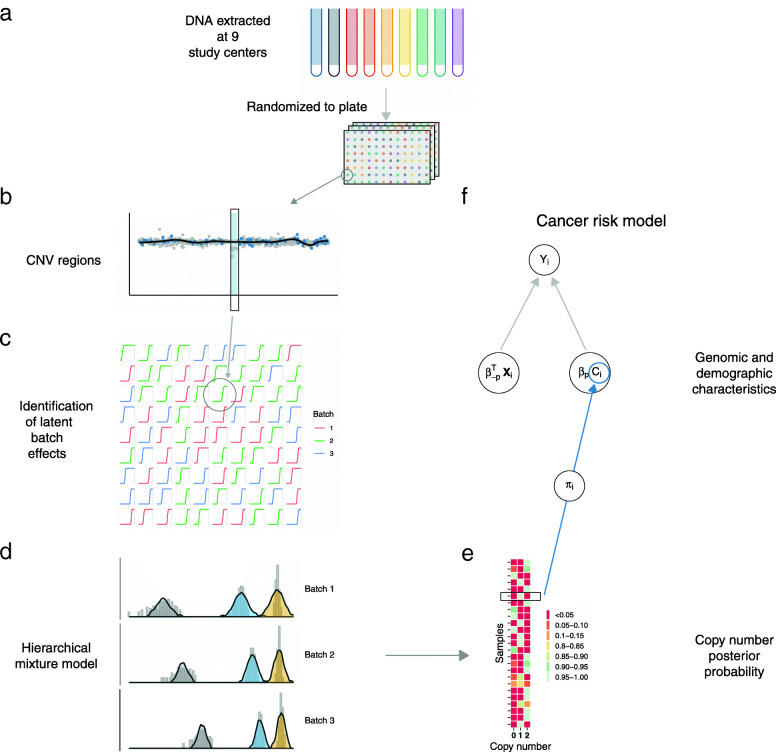
We developed an approach called CNPBayes to identify latent batch effects in genome-wide association studies involving copy number, to provide probabilistic estimates of integer copy number across the estimated batches, and to fully integrate the copy number uncertainty in the association model for disease.
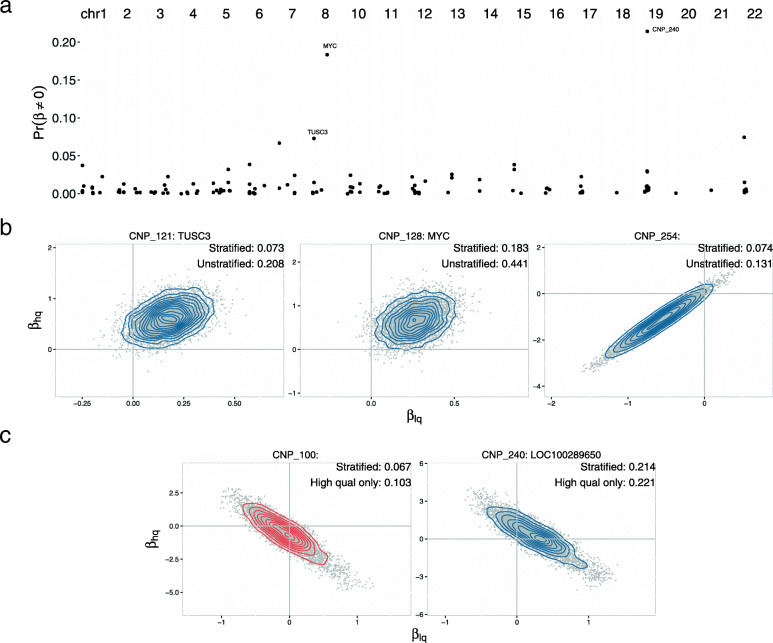
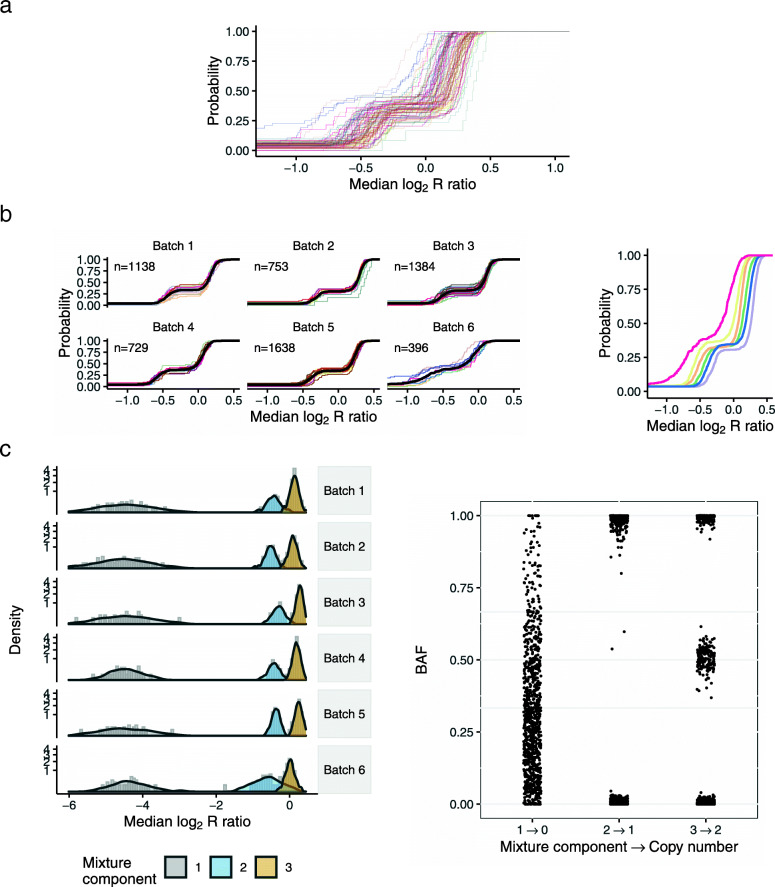
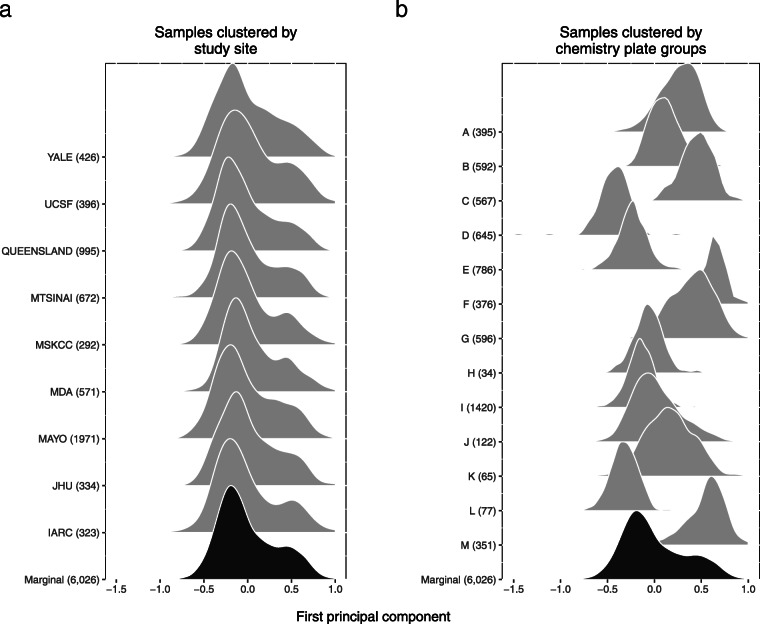
Applying a hidden Markov model (HMM) to identify CNVs in a large multi-site Pancreatic Cancer Case Control study (PanC4) of 7598 participants, we found CNV inference was highly sensitive to technical noise that varied appreciably among participants. Applying CNPBayes to this dataset, we found that the major sources of technical variation were linked to sample processing by the centralized laboratory and not the individual study sites. Modeling the latent batch effects at each CNV region hierarchically, we developed probabilistic estimates of copy number that were directly incorporated in a Bayesian regression model for pancreatic cancer risk. Candidate associations aided by this approach include deletions of 8q24 near regulatory elements of the tumor oncogene MYC and of Tumor Suppressor Candidate 3 (TUSC3).
Laboratory effects may not account for the major sources of technical variation in genome-wide association studies. This study provides a robust Bayesian inferential framework for identifying latent batch effects, estimating copy number, and evaluating the role of copy number in heritable diseases.
种系拷贝数变异 (CNVs) 会增加许多疾病的风险,但由于基因组内和样本间存在生物学和技术异质性,因此在大规模研究中检测 CNVs 并量化其对疾病风险的贡献具有挑战性。
我们开发了一种称为 CNPBayes 的方法,用于识别涉及拷贝数的全基因组关联研究中的潜在批次效应,提供估计批次中整数拷贝数的概率估计,并在关联模型中充分整合拷贝数不确定性以研究疾病。
在一个涉及 7598 名参与者的大型多站点胰腺癌病例对照研究(PanC4)中,我们应用隐马尔可夫模型 (HMM) 识别 CNV,发现 CNV 推断对技术噪声高度敏感,而技术噪声在参与者之间存在明显差异。我们将 CNPBayes 应用于该数据集,发现主要的技术变异源与集中实验室的样本处理有关,而与个别研究地点无关。对每个 CNV 区域的潜在批次效应进行分层建模,我们开发了拷贝数的概率估计值,这些值直接纳入了用于胰腺癌风险的贝叶斯回归模型。通过这种方法辅助的候选关联包括肿瘤致癌基因 MYC 和肿瘤抑制候选 3 (TUSC3) 附近的 8q24 缺失。
实验室效应可能无法解释全基因组关联研究中主要的技术变异源。本研究为识别潜在批次效应、估计拷贝数以及评估拷贝数在遗传性疾病中的作用提供了稳健的贝叶斯推断框架。