Takata Miwako, Kiyohara Akihiro, Takasu Atsuko, Kishima Yuji, Ohtsubo Hisako, Sano Yoshio
Laboratory of Plant Breeding, Graduate School of Agriculture, Hokkaido University, Sapporo 060-8589, Japan.
BMC Genomics. 2007 Dec 20;8:469. doi: 10.1186/1471-2164-8-469.
Recent studies using high-throughput methods have revealed that transposable elements (TEs) are a comprehensive target for DNA methylation. However, the relationship between TEs and their genomic environment regarding methylation still remains unclear. The rice genome contains representatives of all known TE families with different characteristics of chromosomal distribution, structure, transposition, size, and copy number. Here we studied the DNA methylation state around 12 TEs in nine genomic DNAs from cultivated rice strains and their closely related wild strains.
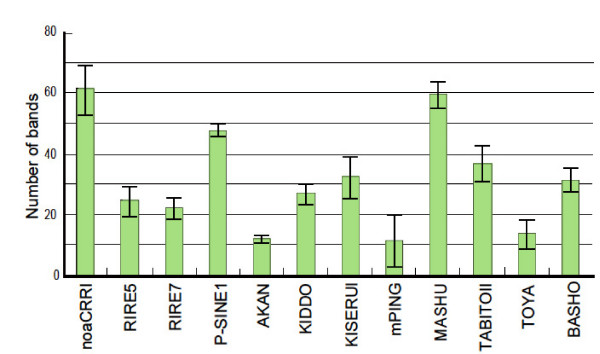
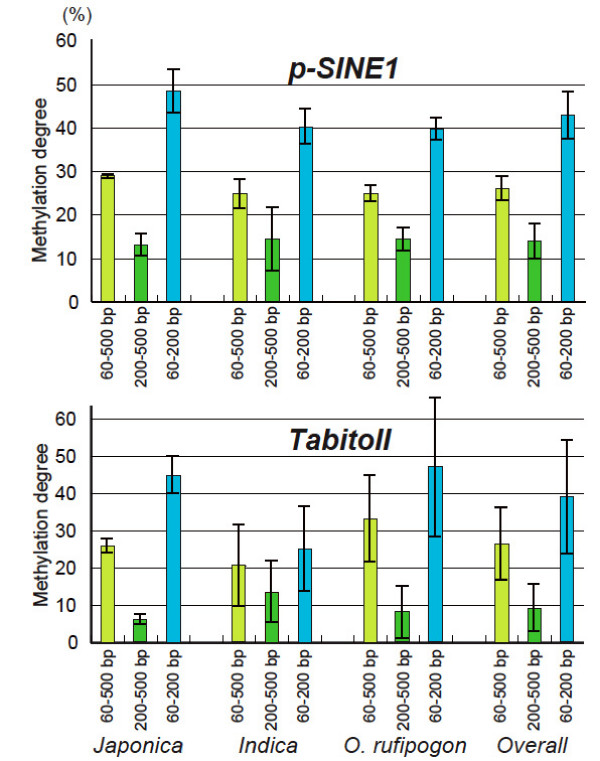
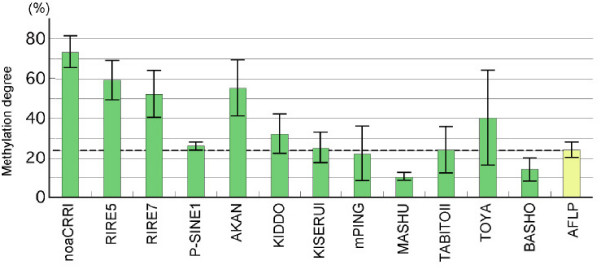
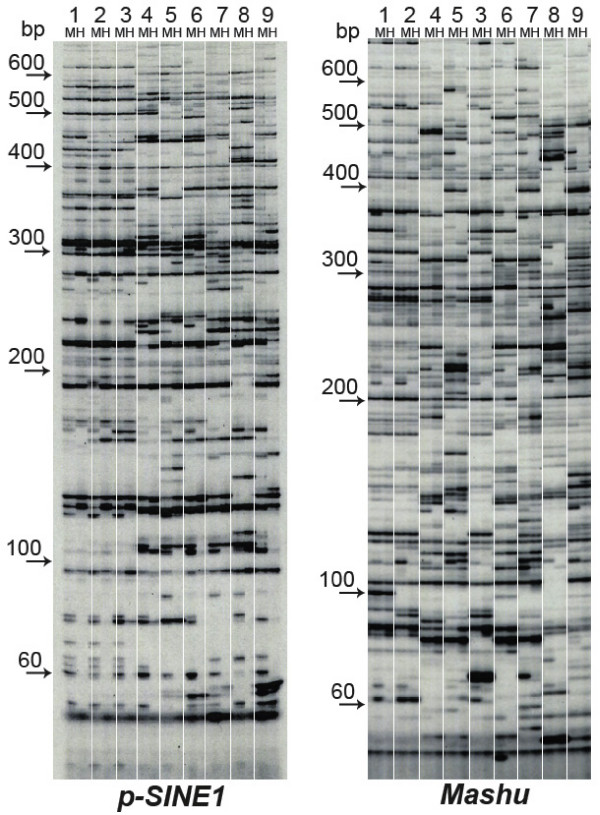
We employed a transposon display (TD) method to analyze the methylation environments in the genomes. The 12 TE families, consisting of four class I elements, seven class II elements, and one element of a different class, were differentially distributed in the rice chromosomes: some elements were concentrated in the centromeric or pericentromeric regions, but others were located in euchromatic regions. The TD analyses revealed that the TE families were embedded in flanking sequences with different methylation degrees. Each TE had flanking sequences with similar degrees of methylation among the nine rice strains. The class I elements tended to be present in highly methylated regions, while those of the class II elements showed widely varying degrees of methylation. In some TE families, the degrees of methylation were markedly lower than the average methylation state of the genome. In two families, dramatic changes of the methylation state occurred depending on the distance from the TE.
Our results demonstrate that the TE families in the rice genomes can be characterized by the methylation states of their surroundings. The copy number and degree of conservation of the TE family are not likely to be correlated with the degree of methylation. We discuss possible relationships between the methylation state of TEs and their surroundings. This is the first report demonstrating that TEs in the genome are associated with a particular methylation environment that is a feature of a given TE.
近期使用高通量方法的研究表明,转座元件(TEs)是DNA甲基化的一个全面靶点。然而,TEs与其基因组环境在甲基化方面的关系仍不清楚。水稻基因组包含了所有已知TE家族的代表,这些家族在染色体分布、结构、转座、大小和拷贝数方面具有不同的特征。在此,我们研究了来自栽培水稻品种及其近缘野生品种的9种基因组DNA中12个TEs周围的DNA甲基化状态。
我们采用转座子展示(TD)方法分析基因组中的甲基化环境。这12个TE家族,包括4个I类元件、7个II类元件和1个不同类别的元件,在水稻染色体上分布不同:一些元件集中在着丝粒或着丝粒周围区域,但其他元件位于常染色质区域。TD分析表明,TE家族嵌入在甲基化程度不同的侧翼序列中。在9个水稻品种中,每个TE都有甲基化程度相似的侧翼序列。I类元件倾向于存在于高度甲基化区域,而II类元件则表现出广泛不同的甲基化程度。在一些TE家族中,甲基化程度明显低于基因组的平均甲基化状态。在两个家族中,甲基化状态根据与TE的距离发生显著变化。
我们的结果表明,水稻基因组中的TE家族可以通过其周围的甲基化状态来表征。TE家族的拷贝数和保守程度不太可能与甲基化程度相关。我们讨论了TEs甲基化状态与其周围环境之间可能的关系。这是第一份证明基因组中的TEs与特定甲基化环境相关的报告,这种环境是特定TE的一个特征。