Mihalek I, Res I, Lichtarge O
Department of Molecular and Human Genetics, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA.
BMC Bioinformatics. 2007 Dec 27;8:488. doi: 10.1186/1471-2105-8-488.
Shannon entropy applied to columns of multiple sequence alignments as a score of residue conservation has proven one of the most fruitful ideas in bioinformatics. This straightforward and intuitively appealing measure clearly shows the regions of a protein under increased evolutionary pressure, highlighting their functional importance. The inability of the column entropy to differentiate between residue types, however, limits its resolution power.
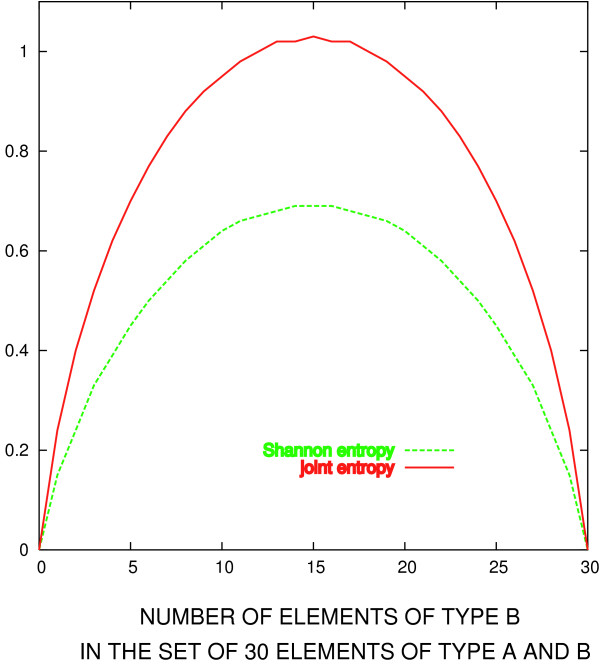
In this work we suggest generalizing Shannon's expression to a function with similar mathematical properties, that, at the same time, includes observed propensities of residue types to mutate to each other. To do that, we revisit the original construction of BLOSUM matrices, and re-interpret them as mutation probability matrices. These probabilities are then used as background frequencies in the revised residue conservation measure.
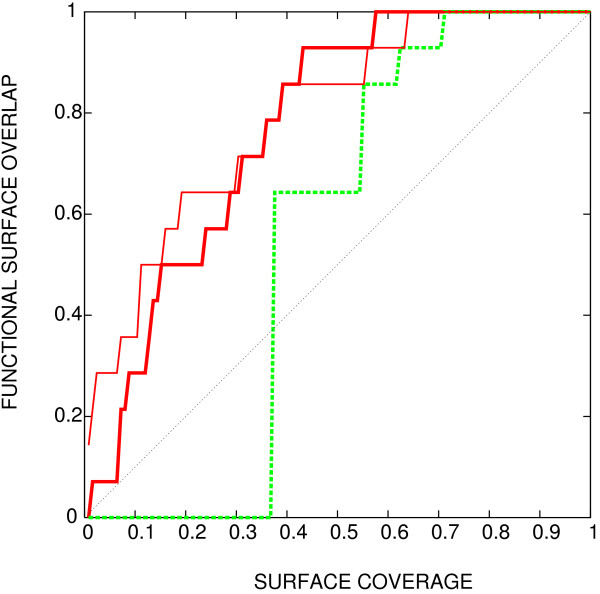
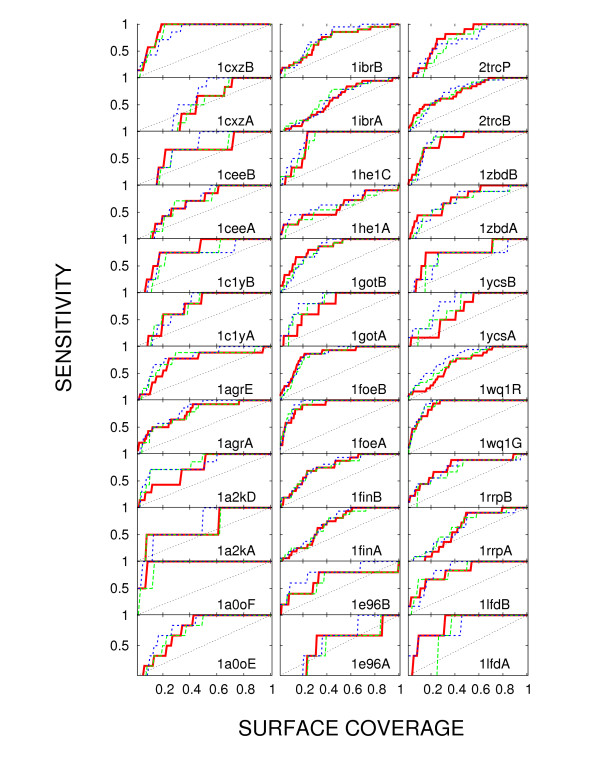
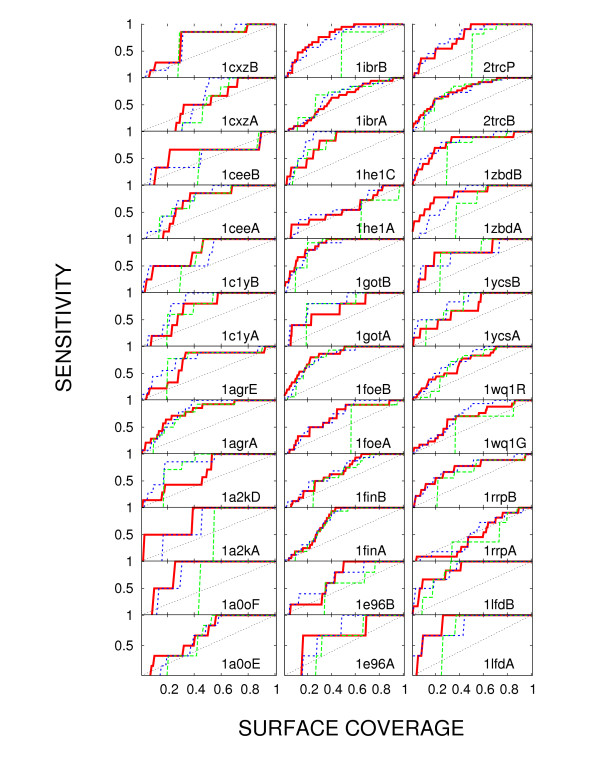
We show that joint entropy with BLOSUM-proportional probabilities as a reference distribution enables detection of protein functional sites comparable in quality to a time-costly maximum-likelihood evolution simulation method (rate4site), and offers greater resolution than the Shannon entropy alone, in particular in the cases when the available sequences are of narrow evolutionary scope.
将香农熵应用于多序列比对的列,作为残基保守性的评分,已被证明是生物信息学中最富有成效的理念之一。这种直接且直观的吸引人的度量方法清楚地显示了蛋白质在进化压力增加下的区域,突出了它们的功能重要性。然而,列熵无法区分残基类型,这限制了它的分辨能力。
在这项工作中,我们建议将香农表达式推广到具有相似数学性质的函数,该函数同时包含观察到的残基类型相互突变的倾向。为此,我们重新审视BLOSUM矩阵的原始构建,并将它们重新解释为突变概率矩阵。然后,这些概率被用作修订后的残基保守性度量中的背景频率。
我们表明,以BLOSUM比例概率作为参考分布的联合熵能够检测出与耗时的最大似然进化模拟方法(rate4site)质量相当的蛋白质功能位点,并且比单独的香农熵具有更高的分辨率,特别是在可用序列的进化范围较窄的情况下。