Watkinson John, Wang Xiaodong, Zheng Tian, Anastassiou Dimitris
Center for Computational Biology and Bioinformatics and Department of Electrical Engineering, Columbia University, 500 West 120th Street, New York, NY 10027, USA.
BMC Syst Biol. 2008 Jan 30;2:10. doi: 10.1186/1752-0509-2-10.
Analysis of microarray data has been used for the inference of gene-gene interactions. If, however, the aim is the discovery of disease-related biological mechanisms, then the criterion for defining such interactions must be specifically linked to disease.
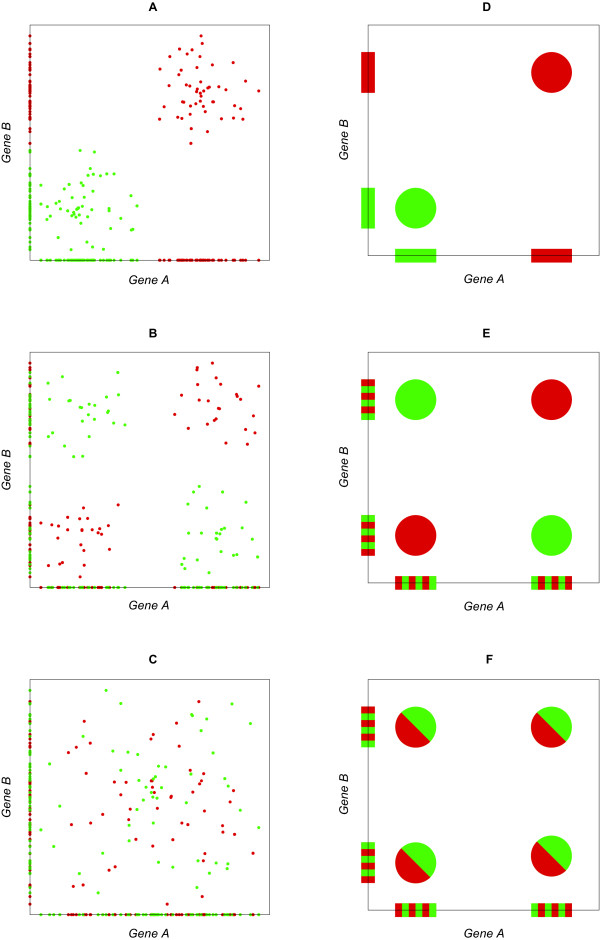
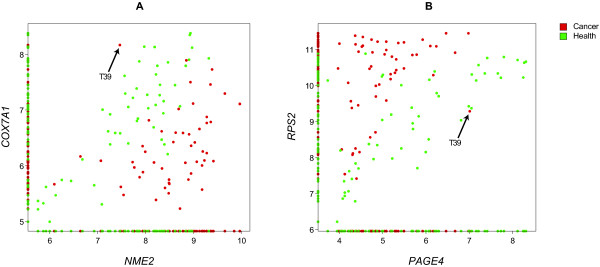
Here we present a computational methodology that jointly analyzes two sets of microarray data, one in the presence and one in the absence of a disease, identifying gene pairs whose correlation with disease is due to cooperative, rather than independent, contributions of genes, using the recently developed information theoretic measure of synergy. High levels of synergy in gene pairs indicates possible membership of the two genes in a shared pathway and leads to a graphical representation of inferred gene-gene interactions associated with disease, in the form of a "synergy network." We apply this technique on a set of publicly available prostate cancer expression data and successfully validate our results, confirming that they cannot be due to pure chance and providing a biological explanation for gene pairs with exceptionally high synergy.
Thus, synergy networks provide a computational methodology helpful for deriving "disease interactomes" from biological data. When coupled with additional biological knowledge, they can also be helpful for deciphering biological mechanisms responsible for disease.
微阵列数据分析已被用于推断基因-基因相互作用。然而,如果目标是发现与疾病相关的生物学机制,那么定义此类相互作用的标准必须与疾病有特定关联。
在此,我们提出一种计算方法,该方法联合分析两组微阵列数据,一组是疾病存在时的数据,另一组是疾病不存在时的数据,使用最近开发的协同信息理论度量来识别那些基因对,其与疾病的相关性是由于基因的协同而非独立作用。基因对中的高协同水平表明这两个基因可能属于共同途径,并导致以“协同网络”形式呈现与疾病相关的推断基因-基因相互作用的图形表示。我们将此技术应用于一组公开可用的前列腺癌表达数据,并成功验证了我们的结果,确认它们并非纯粹出于偶然,并为具有异常高协同性的基因对提供了生物学解释。
因此,协同网络提供了一种有助于从生物学数据中推导“疾病相互作用组”的计算方法。当与其他生物学知识相结合时,它们也有助于解读导致疾病的生物学机制。