Yang Kuan, Zhang Liqing
Virginia Bioinformatics Institute, Virginia, USA.
Nucleic Acids Res. 2008 Mar;36(5):e33. doi: 10.1093/nar/gkn075. Epub 2008 Feb 22.
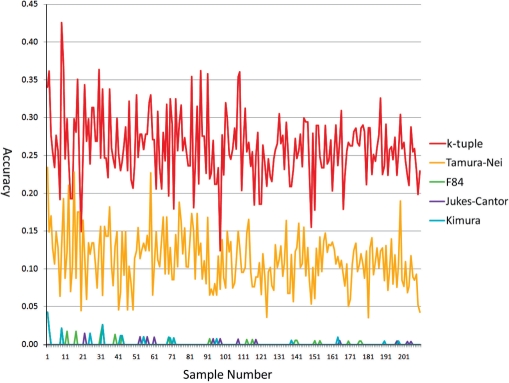
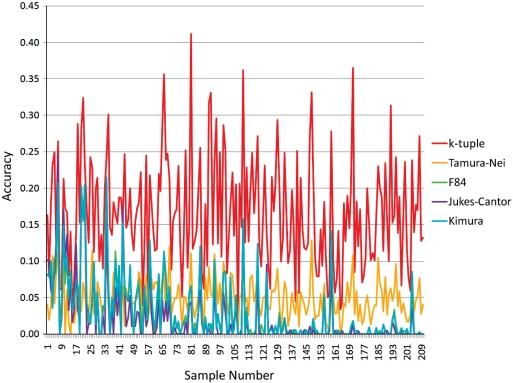
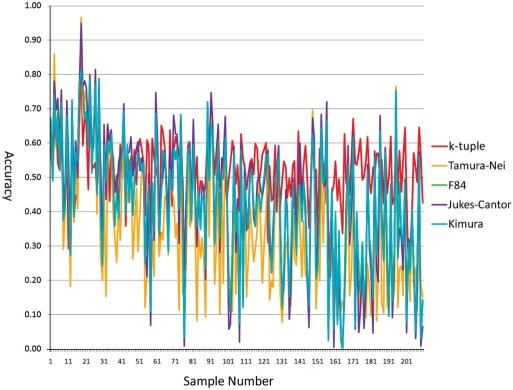
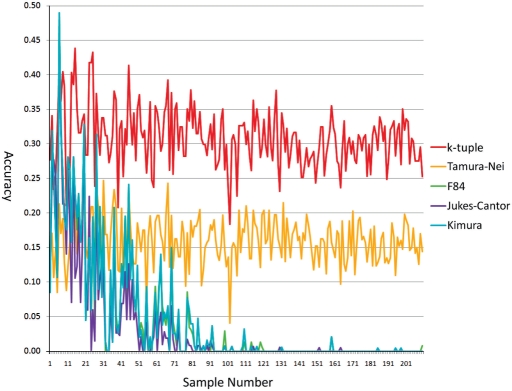
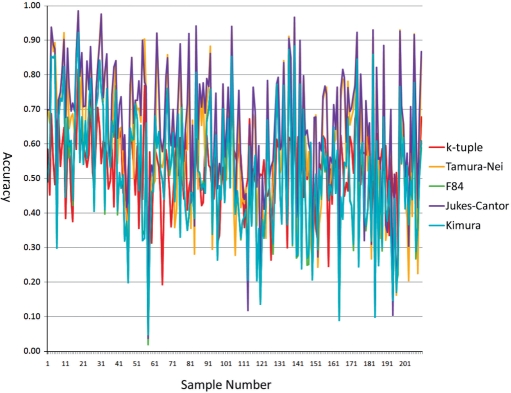
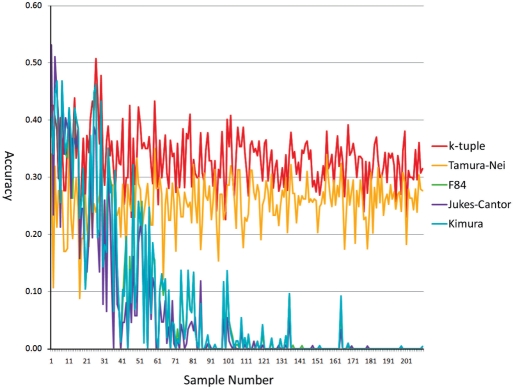
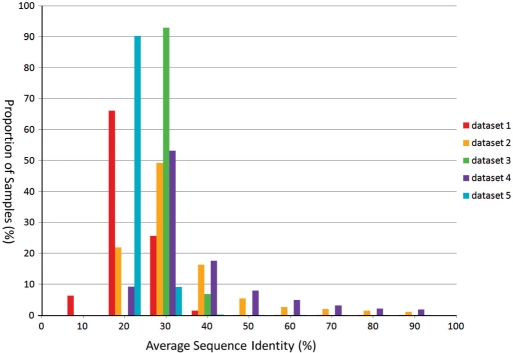
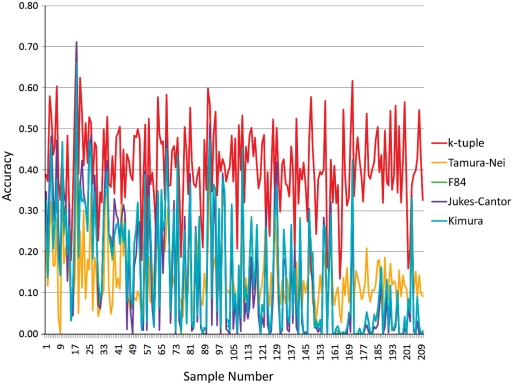
Phylogenetic tree reconstruction requires construction of a multiple sequence alignment (MSA) from sequences. Computationally, it is difficult to achieve an optimal MSA for many sequences. Moreover, even if an optimal MSA is obtained, it may not be the true MSA that reflects the evolutionary history of the underlying sequences. Therefore, errors can be introduced during MSA construction which in turn affects the subsequent phylogenetic tree construction. In order to circumvent this issue, we extend the application of the k-tuple distance to phylogenetic tree reconstruction. The k-tuple distance between two sequences is the sum of the differences in frequency, over all possible tuples of length k, between the sequences and can be estimated without MSAs. It has been traditionally used to build a fast 'guide tree' to assist the construction of MSAs. Using the 1470 simulated sets of sequences generated under different evolutionary scenarios, the neighbor-joining trees and BioNJ trees, we compared the performance of the k-tuple distance with four commonly used distance estimators including Jukes-Cantor, Kimura, F84 and Tamura-Nei. These four distance estimators fall into the category of model-based distance estimators, as each of them takes account of a specific substitution model in order to compute the distance between a pair of already aligned sequences. Results show that trees constructed from the k-tuple distance are more accurate than those from other distances most time; when the divergence between underlying sequences is high, the tree accuracy could be twice or higher using the k-tuple distance than other estimators. Furthermore, as the k-tuple distance voids the need for constructing an MSA, it can save tremendous amount of time for phylogenetic tree reconstructions when the data include a large number of sequences.
系统发育树重建需要从序列构建多序列比对(MSA)。从计算角度来看,为许多序列获得最优的MSA是困难的。此外,即使获得了最优的MSA,它可能也不是反映基础序列进化历史的真实MSA。因此,在MSA构建过程中可能会引入误差,这进而会影响后续的系统发育树构建。为了规避这个问题,我们将k元组距离的应用扩展到系统发育树重建。两个序列之间的k元组距离是在所有长度为k的可能元组上,序列之间频率差异的总和,并且无需MSA即可估计。传统上它被用于构建快速的“引导树”以辅助MSA的构建。使用在不同进化场景下生成的1470组模拟序列、邻接法树和BioNJ树,我们将k元组距离的性能与四种常用的距离估计器进行了比较,包括Jukes-Cantor、Kimura、F84和Tamura-Nei。这四种距离估计器属于基于模型的距离估计器类别,因为它们每一个都考虑了特定的替换模型,以便计算一对已比对序列之间的距离。结果表明,在大多数情况下,由k元组距离构建的树比由其他距离构建的树更准确;当基础序列之间的分歧度较高时,使用k元组距离构建的树的准确性可能是其他估计器的两倍或更高。此外,由于k元组距离无需构建MSA,当数据包含大量序列时,它可以为系统发育树重建节省大量时间。