Lähdesmäki Harri, Rust Alistair G, Shmulevich Ilya
Institute for Systems Biology, Seattle, Washington, United States of America.
PLoS One. 2008 Mar 26;3(3):e1820. doi: 10.1371/journal.pone.0001820.
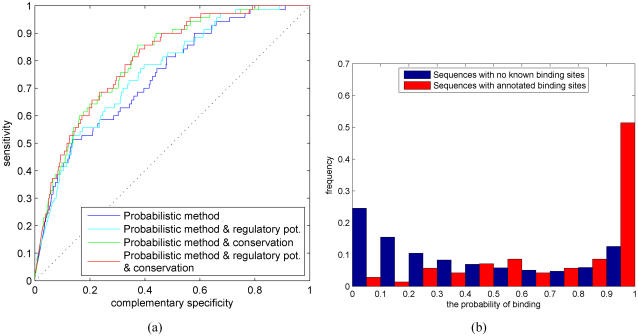
An important problem in molecular biology is to build a complete understanding of transcriptional regulatory processes in the cell. We have developed a flexible, probabilistic framework to predict TF binding from multiple data sources that differs from the standard hypothesis testing (scanning) methods in several ways. Our probabilistic modeling framework estimates the probability of binding and, thus, naturally reflects our degree of belief in binding. Probabilistic modeling also allows for easy and systematic integration of our binding predictions into other probabilistic modeling methods, such as expression-based gene network inference. The method answers the question of whether the whole analyzed promoter has a binding site, but can also be extended to estimate the binding probability at each nucleotide position. Further, we introduce an extension to model combinatorial regulation by several TFs. Most importantly, the proposed methods can make principled probabilistic inference from multiple evidence sources, such as, multiple statistical models (motifs) of the TFs, evolutionary conservation, regulatory potential, CpG islands, nucleosome positioning, DNase hypersensitive sites, ChIP-chip binding segments and other (prior) sequence-based biological knowledge. We developed both a likelihood and a Bayesian method, where the latter is implemented with a Markov chain Monte Carlo algorithm. Results on a carefully constructed test set from the mouse genome demonstrate that principled data fusion can significantly improve the performance of TF binding prediction methods. We also applied the probabilistic modeling framework to all promoters in the mouse genome and the results indicate a sparse connectivity between transcriptional regulators and their target promoters. To facilitate analysis of other sequences and additional data, we have developed an on-line web tool, ProbTF, which implements our probabilistic TF binding prediction method using multiple data sources. Test data set, a web tool, source codes and supplementary data are available at: http://www.probtf.org.
分子生物学中的一个重要问题是全面了解细胞中的转录调控过程。我们开发了一个灵活的概率框架,用于从多个数据源预测转录因子(TF)结合,该框架在几个方面不同于标准的假设检验(扫描)方法。我们的概率建模框架估计结合概率,因此自然地反映了我们对结合的置信程度。概率建模还允许将我们的结合预测轻松、系统地整合到其他概率建模方法中,例如基于表达的基因网络推断。该方法回答了整个分析的启动子是否具有结合位点的问题,但也可以扩展以估计每个核苷酸位置的结合概率。此外,我们引入了一种扩展,用于对多个转录因子的组合调控进行建模。最重要的是,所提出的方法可以从多个证据来源进行有原则的概率推断,例如转录因子的多个统计模型(模体)、进化保守性、调控潜力、CpG岛、核小体定位、DNase超敏位点、ChIP-chip结合片段以及其他(先验)基于序列的生物学知识。我们开发了似然法和贝叶斯方法,后者通过马尔可夫链蒙特卡罗算法实现。来自小鼠基因组的精心构建的测试集上的结果表明,有原则的数据融合可以显著提高转录因子结合预测方法的性能。我们还将概率建模框架应用于小鼠基因组中的所有启动子,结果表明转录调节因子与其靶启动子之间的连接稀疏。为了便于分析其他序列和更多数据,我们开发了一个在线网络工具ProbTF,它使用多个数据源实现了我们的概率转录因子结合预测方法。测试数据集、网络工具、源代码和补充数据可在以下网址获取:http://www.probtf.org。