Yang Tzu-Hsien, Wu Wei-Sheng
BMC Syst Biol. 2013;7 Suppl 6(Suppl 6):S13. doi: 10.1186/1752-0509-7-S6-S13. Epub 2013 Dec 13.
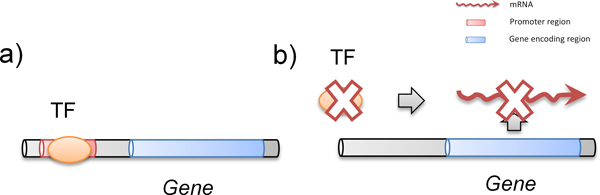
Chromatin immunoprecipitation (ChIP) experiments are now the most comprehensive experimental approaches for mapping the binding of transcription factors (TFs) to their target genes. However, ChIP data alone is insufficient for identifying functional binding target genes of TFs for two reasons. First, there is an inherent high false positive/negative rate in ChIP-chip or ChIP-seq experiments. Second, binding signals in the ChIP data do not necessarily imply functionality.
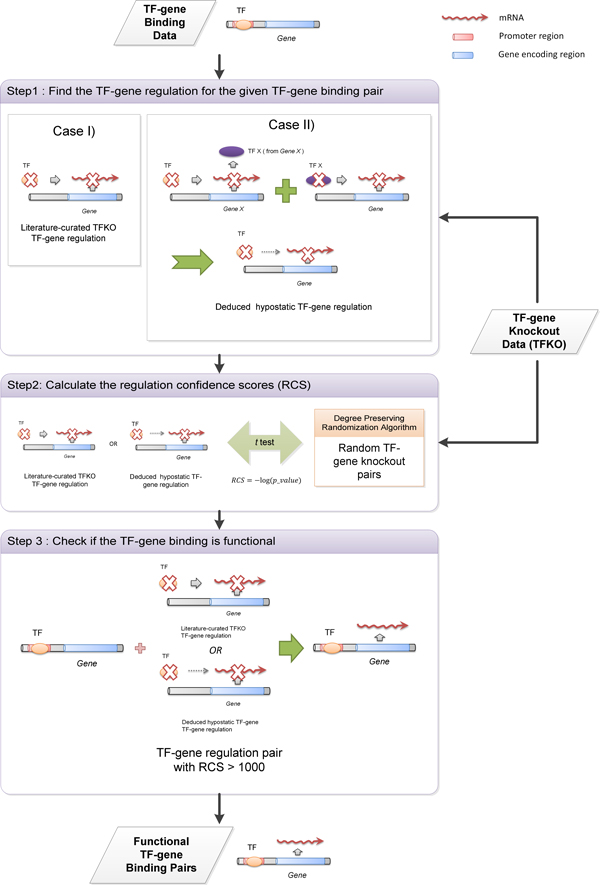
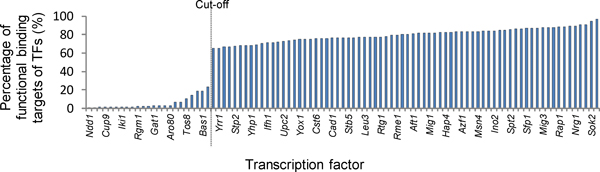
It is known that ChIP-chip data and TF knockout (TFKO) data reveal complementary information on gene regulation. While ChIP-chip data can provide TF-gene binding pairs, TFKO data can provide TF-gene regulation pairs. Therefore, we propose a novel network approach for identifying functional TF-gene binding pairs by integrating the ChIP-chip data with the TFKO data. In our method, a TF-gene binding pair from the ChIP-chip data is regarded to be functional if it also has high confident curated TFKO TF-gene regulatory relation or deduced hypostatic TF-gene regulatory relation.
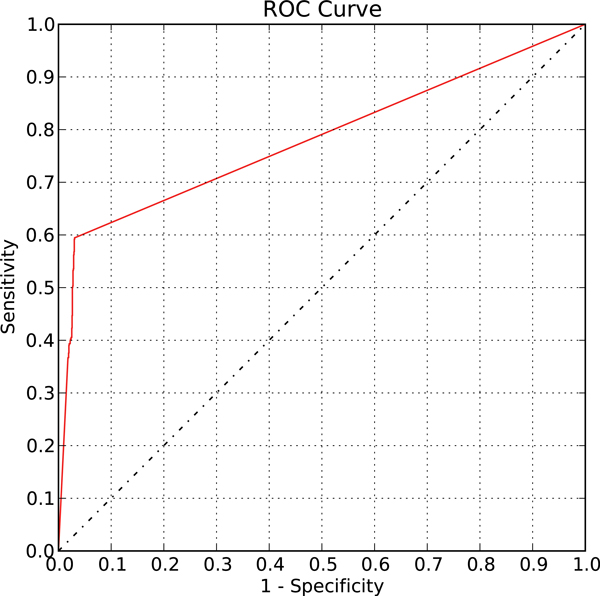
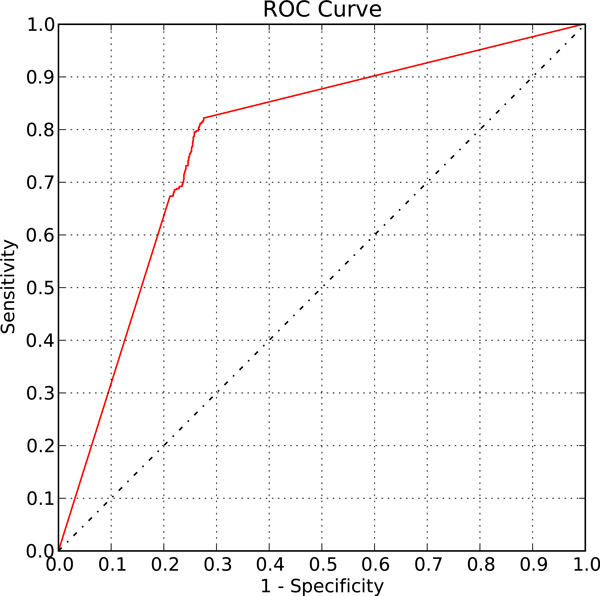
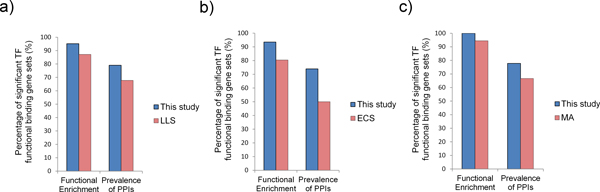
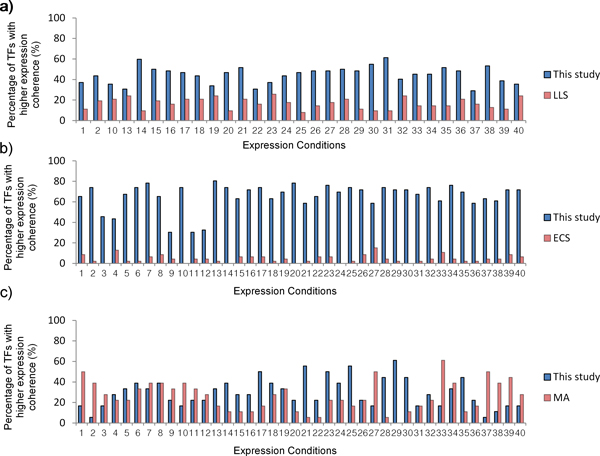
We first validated our method on a gathered ground truth set. Then we applied our method to the ChIP-chip data to identify functional TF-gene binding pairs. The biological significance of our identified functional TF-gene binding pairs was shown by assessing their functional enrichment, the prevalence of protein-protein interaction, and expression coherence. Our results outperformed the results of three existing methods across all measures. And our identified functional targets of TFs also showed statistical significance over the randomly assigned TF-gene pairs. We also showed that our method is dataset independent and can apply to ChIP-seq data and the E. coli genome. Finally, we provided an example showing the biological applicability of our notion.
染色质免疫沉淀(ChIP)实验是目前用于绘制转录因子(TFs)与其靶基因结合图谱的最全面的实验方法。然而,仅ChIP数据不足以识别TFs的功能性结合靶基因,原因有两个。第一,ChIP芯片或ChIP测序实验中存在固有的高假阳性/阴性率。第二,ChIP数据中的结合信号不一定意味着功能性。
已知ChIP芯片数据和TF敲除(TFKO)数据揭示了关于基因调控的互补信息。虽然ChIP芯片数据可以提供TF-基因结合对,但TFKO数据可以提供TF-基因调控对。因此,我们提出了一种新的网络方法,通过将ChIP芯片数据与TFKO数据整合来识别功能性TF-基因结合对。在我们的方法中,如果来自ChIP芯片数据的TF-基因结合对也具有高可信度的经过整理的TFKO TF-基因调控关系或推导的下位TF-基因调控关系,则认为它是功能性的。
我们首先在一个收集的真值集上验证了我们的方法。然后我们将我们的方法应用于ChIP芯片数据以识别功能性TF-基因结合对。通过评估它们的功能富集、蛋白质-蛋白质相互作用的普遍性和表达一致性,显示了我们识别的功能性TF-基因结合对的生物学意义。在所有指标上,我们的结果都优于三种现有方法的结果。而且我们识别的TFs的功能性靶标在随机分配的TF-基因对中也显示出统计学意义。我们还表明我们的方法与数据集无关,并且可以应用于ChIP测序数据和大肠杆菌基因组。最后,我们提供了一个例子来说明我们概念的生物学适用性。