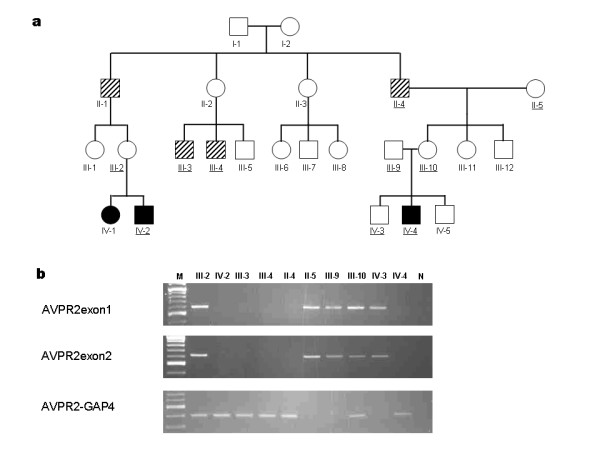
Fujimoto Masaya, Imai Kohsuke, Hirata Kenji, Kashiwagi Reiichi, Morinishi Yoichi, Kitazawa Katsuhiko, Sasaki Sei, Arinami Tadao, Nonoyama Shigeaki, Noguchi Emiko
Department of Medical Genetics, Graduate School of Comprehensive Human Sciences, University of Tsukuba, Ibaraki, Japan.
BMC Med Genet. 2008 May 20;9:42. doi: 10.1186/1471-2350-9-42.
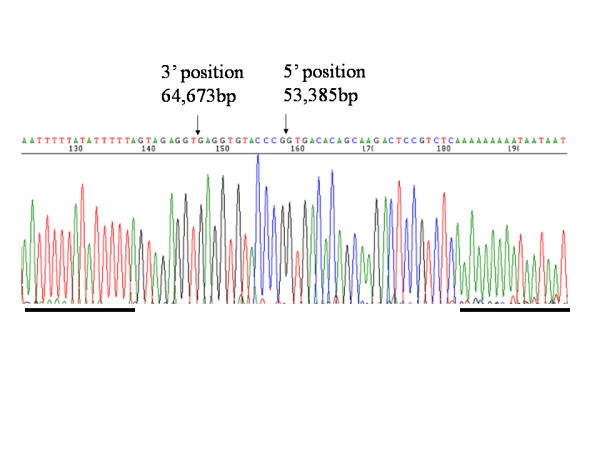
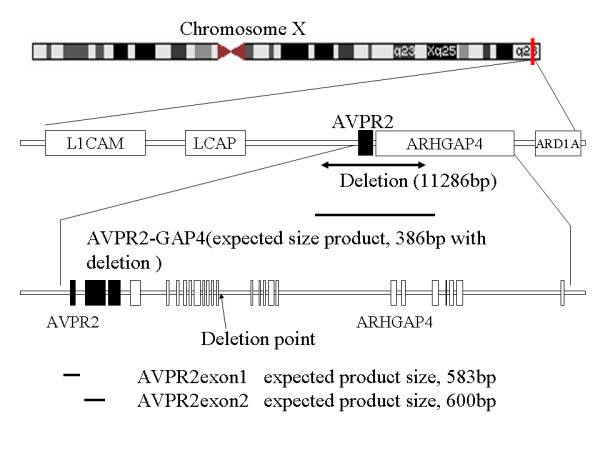
Congenital nephrogenic diabetes insipidus (NDI) is characterised by an inability to concentrate urine despite normal or elevated plasma levels of the antidiuretic hormone arginine vasopressin. We report a Japanese extended family with NDI caused by an 11.2-kb deletion that includes the entire AVPR2 locus and approximately half of the Rho GTPase-activating protein 4 (ARHGAP4) locus. ARHGAP4 belongs to the RhoGAP family, Rho GTPases are critical regulators of many cellular activities, such as motility and proliferation which enhances intrinsic GTPase activity.ARHGAP4 is expressed at high levels in hematopoietic cells, and it has been reported that an NDI patient lacking AVPR2 and all of ARHGAP4 showed immunodeficiency characterised by a marked reduction in the number of circulating CD3+ cells and almost complete absence of CD8+ cells.
PCR and sequencing were performed to identify the deleted region in the Japanese NDI patients. Immunological profiles of the NDI patients were analysed by flow cytometry. We also investigated the gene expression profiles of peripheral blood mononuclear cells (PBMC) from NDI patients and healthy controls in microarray technique.
We evaluated subjects (one child and two adults) with 11.2-kb deletion that includes the entire AVPR2 locus and approximately half of the ARHGAP4. Hematologic tests showed a reduction of CD4+ cells in one adult patient, a reduction in CD8+ cells in the paediatric patient, and a slight reduction in the serum IgG levels in the adult patients, but none of them showed susceptibility to infection. Gene expression profiling of PBMC lacking ARHGAP4 revealed that expression of RhoGAP family genes was not influenced greatly by the lack of ARHGAP4.
These results suggest that loss of ARHGAP4 expression is not compensated for by other family members. ARHGAP4 may play some role in lymphocyte differentiation but partial loss of ARHGAP4 does not result in clinical immunodeficiency.
先天性肾性尿崩症(NDI)的特征是尽管抗利尿激素精氨酸加压素的血浆水平正常或升高,但仍无法浓缩尿液。我们报告了一个患有NDI的日本大家庭,该病由一个11.2kb的缺失引起,该缺失包括整个AVPR2基因座和约一半的Rho GTP酶激活蛋白4(ARHGAP4)基因座。ARHGAP4属于RhoGAP家族,Rho GTP酶是许多细胞活动的关键调节因子,如运动和增殖,可增强内在GTP酶活性。ARHGAP4在造血细胞中高水平表达,据报道,一名缺乏AVPR2和所有ARHGAP4的NDI患者表现出免疫缺陷,其特征是循环CD3 +细胞数量显著减少,几乎完全没有CD8 +细胞。
进行PCR和测序以鉴定日本NDI患者中的缺失区域。通过流式细胞术分析NDI患者的免疫谱。我们还采用微阵列技术研究了NDI患者和健康对照外周血单个核细胞(PBMC)的基因表达谱。
我们评估了具有11.2kb缺失的受试者(一名儿童和两名成人),该缺失包括整个AVPR2基因座和约一半的ARHGAP4。血液学检查显示一名成年患者的CD4 +细胞减少,儿科患者的CD8 +细胞减少,成年患者的血清IgG水平略有降低,但他们均未表现出感染易感性。缺乏ARHGAP4的PBMC的基因表达谱分析显示,RhoGAP家族基因的表达并未受到ARHGAP4缺失的很大影响。
这些结果表明,ARHGAP4表达的缺失不能由其他家族成员代偿。ARHGAP4可能在淋巴细胞分化中起某些作用,但ARHGAP4的部分缺失不会导致临床免疫缺陷。