Espadaler Jordi, Eswar Narayanan, Querol Enrique, Avilés Francesc X, Sali Andrej, Marti-Renom Marc A, Oliva Baldomero
Laboratori de Bioinformàtica Estructural (GRIB), Departament de Ciències Experimentals i de la Salut, Universitat Pompeu Fabra-IMIM, 08003-Barcelona, Catalonia, Spain.
BMC Bioinformatics. 2008 May 27;9:249. doi: 10.1186/1471-2105-9-249.
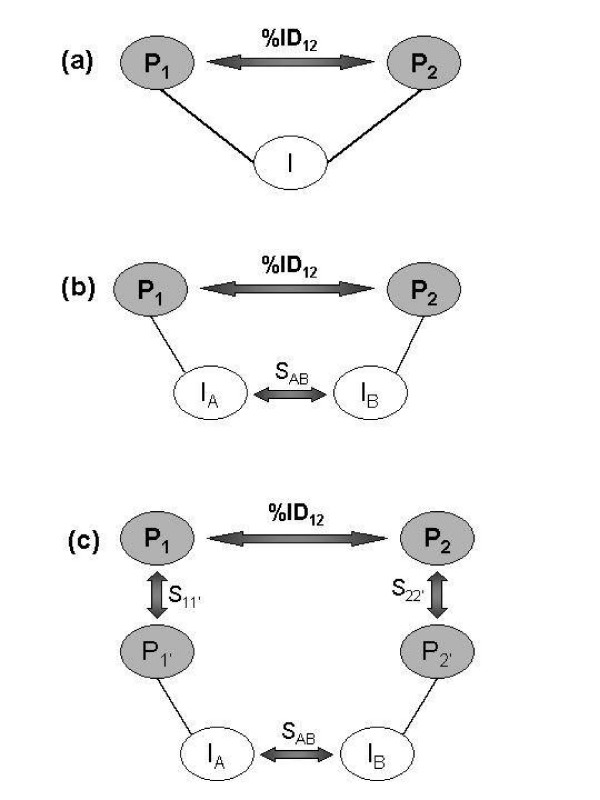
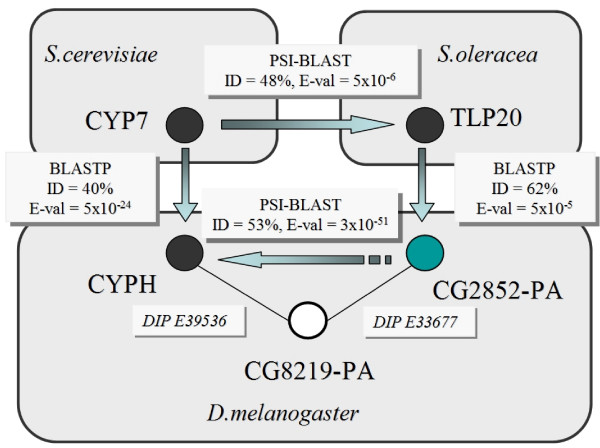
A number of studies have used protein interaction data alone for protein function prediction. Here, we introduce a computational approach for annotation of enzymes, based on the observation that similar protein sequences are more likely to perform the same function if they share similar interacting partners.
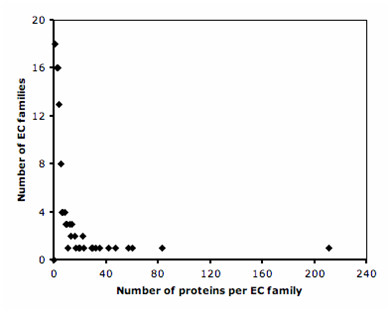
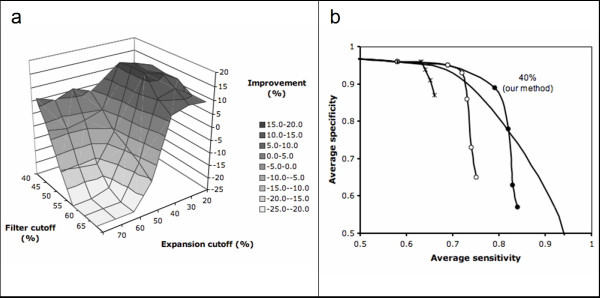
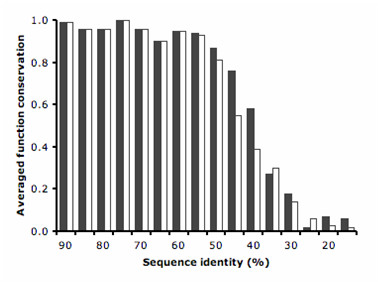
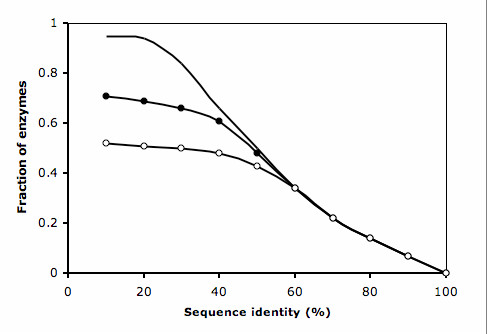
The method has been tested against the PSI-BLAST program using a set of 3,890 protein sequences from which interaction data was available. For protein sequences that align with at least 40% sequence identity to a known enzyme, the specificity of our method in predicting the first three EC digits increased from 80% to 90% at 80% coverage when compared to PSI-BLAST.
Our method can also be used in proteins for which homologous sequences with known interacting partners can be detected. Thus, our method could increase 10% the specificity of genome-wide enzyme predictions based on sequence matching by PSI-BLAST alone.
许多研究仅使用蛋白质相互作用数据来预测蛋白质功能。在此,我们基于这样的观察结果引入了一种用于酶注释的计算方法,即如果相似的蛋白质序列共享相似的相互作用伙伴,则它们更有可能执行相同的功能。
该方法已使用一组3890个可获得相互作用数据的蛋白质序列与PSI-BLAST程序进行了测试。对于与已知酶的序列同一性至少为40%的蛋白质序列,与PSI-BLAST相比,在80%的覆盖率下,我们的方法预测前三个酶委员会(EC)数字的特异性从80%提高到了90%。
我们的方法也可用于能够检测到具有已知相互作用伙伴的同源序列的蛋白质。因此,我们的方法可以使仅基于PSI-BLAST序列匹配的全基因组酶预测的特异性提高10%。