Chan Timothy A, Glockner Sabine, Yi Joo Mi, Chen Wei, Van Neste Leander, Cope Leslie, Herman James G, Velculescu Victor, Schuebel Kornel E, Ahuja Nita, Baylin Stephen B
Cancer Biology Program, The Johns Hopkins Kimmel Cancer Center, Baltimore, Maryland, United States of America.
PLoS Med. 2008 May 27;5(5):e114. doi: 10.1371/journal.pmed.0050114.
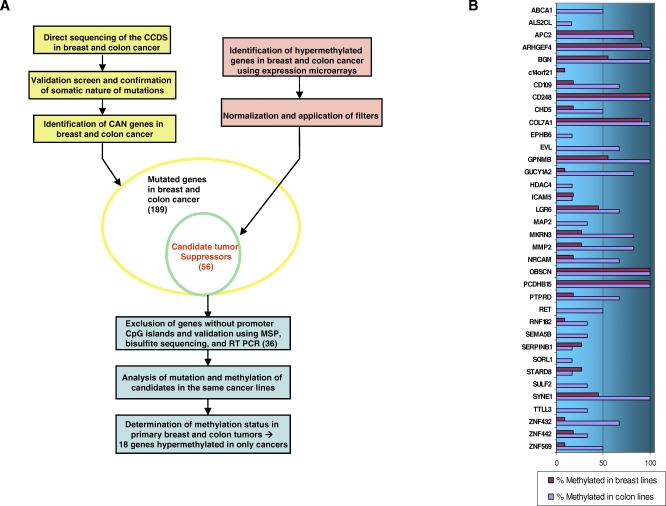
The identification and characterization of tumor suppressor genes has enhanced our understanding of the biology of cancer and enabled the development of new diagnostic and therapeutic modalities. Whereas in past decades, a handful of tumor suppressors have been slowly identified using techniques such as linkage analysis, large-scale sequencing of the cancer genome has enabled the rapid identification of a large number of genes that are mutated in cancer. However, determining which of these many genes play key roles in cancer development has proven challenging. Specifically, recent sequencing of human breast and colon cancers has revealed a large number of somatic gene mutations, but virtually all are heterozygous, occur at low frequency, and are tumor-type specific. We hypothesize that key tumor suppressor genes in cancer may be subject to mutation or hypermethylation.
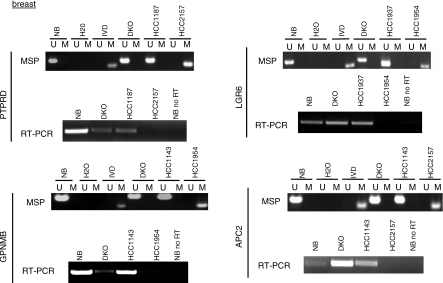
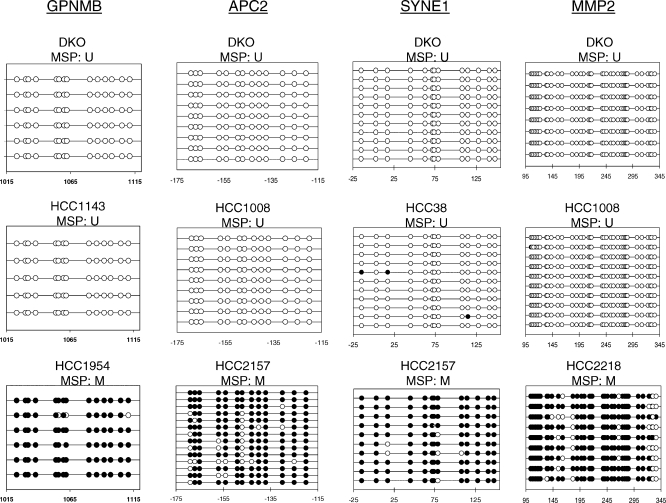
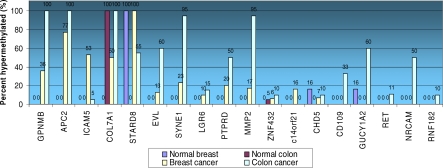
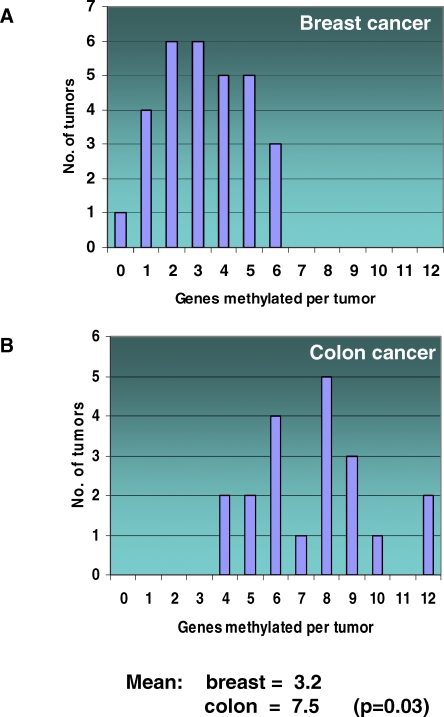
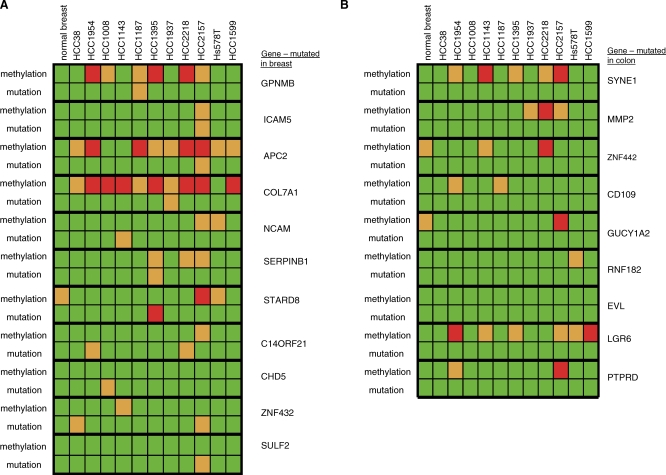
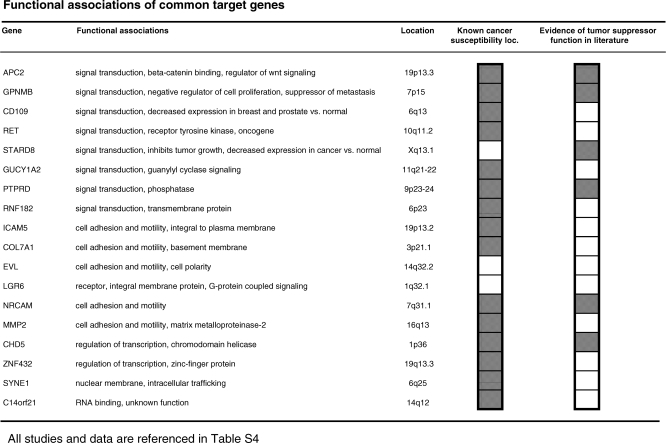
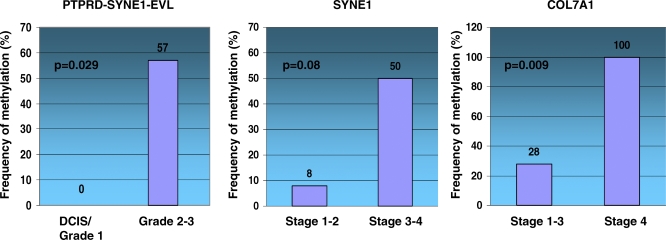
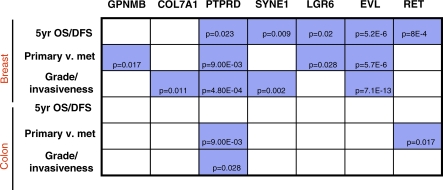
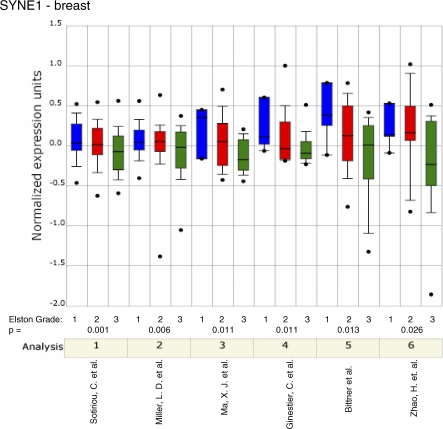
Here, we show that combined genetic and epigenetic analysis of these genes reveals many with a higher putative tumor suppressor status than would otherwise be appreciated. At least 36 of the 189 genes newly recognized to be mutated are targets of promoter CpG island hypermethylation, often in both colon and breast cancer cell lines. Analyses of primary tumors show that 18 of these genes are hypermethylated strictly in primary cancers and often with an incidence that is much higher than for the mutations and which is not restricted to a single tumor-type. In the identical breast cancer cell lines in which the mutations were identified, hypermethylation is usually, but not always, mutually exclusive from genetic changes for a given tumor, and there is a high incidence of concomitant loss of expression. Sixteen out of 18 (89%) of these genes map to loci deleted in human cancers. Lastly, and most importantly, the reduced expression of a subset of these genes strongly correlates with poor clinical outcome.
Using an unbiased genome-wide approach, our analysis has enabled the discovery of a number of clinically significant genes targeted by multiple modes of inactivation in breast and colon cancer. Importantly, we demonstrate that a subset of these genes predict strongly for poor clinical outcome. Our data define a set of genes that are targeted by both genetic and epigenetic events, predict for clinical prognosis, and are likely fundamentally important for cancer initiation or progression.
肿瘤抑制基因的鉴定和特征分析增进了我们对癌症生物学的理解,并推动了新诊断和治疗方法的发展。在过去几十年中,通过连锁分析等技术缓慢鉴定出了少数肿瘤抑制基因,而癌症基因组的大规模测序使得快速鉴定出大量在癌症中发生突变的基因成为可能。然而,确定这些众多基因中哪些在癌症发展中起关键作用已被证明具有挑战性。具体而言,最近对人类乳腺癌和结肠癌的测序揭示了大量体细胞基因突变,但几乎所有突变都是杂合的,发生频率低,且具有肿瘤类型特异性。我们推测癌症中的关键肿瘤抑制基因可能会发生突变或高甲基化。
在这里,我们表明对这些基因进行联合遗传和表观遗传分析发现,许多基因具有比其他情况更高的假定肿瘤抑制状态。新确认发生突变的189个基因中至少有36个是启动子CpG岛高甲基化的靶点,在结肠和乳腺癌细胞系中通常都是如此。对原发性肿瘤的分析表明,这些基因中有18个在原发性癌症中严格发生高甲基化,其发生率通常远高于突变发生率,且不限于单一肿瘤类型。在鉴定出突变的相同乳腺癌细胞系中,高甲基化通常(但并非总是)与给定肿瘤的基因变化相互排斥,并且存在表达同时缺失的高发生率。这些基因中有18个中的16个(89%)定位于人类癌症中缺失的位点。最后,也是最重要的,这些基因中的一部分表达降低与不良临床结果密切相关。
通过无偏倚的全基因组方法,我们的分析发现了一些在乳腺癌和结肠癌中受到多种失活模式影响的具有临床意义的基因。重要的是,我们证明这些基因中的一部分对不良临床结果有很强的预测作用。我们的数据定义了一组受遗传和表观遗传事件影响、预测临床预后且可能对癌症起始或进展至关重要的基因。