Nakayama Keisuke, Yamashita Atsushi, Kurokawa Ken, Morimoto Takuya, Ogawa Michihiro, Fukuhara Masahiro, Urakami Hiroshi, Ohnishi Makoto, Uchiyama Ikuo, Ogura Yoshitoshi, Ooka Tadasuke, Oshima Kenshiro, Tamura Akira, Hattori Masahira, Hayashi Tetsuya
Division of Microbiology, Department of Infectious Diseases, Faculty of Medicine, University of Miyazaki, 5200 Kiyotake, Miyazaki, Japan.
DNA Res. 2008 Aug;15(4):185-99. doi: 10.1093/dnares/dsn011. Epub 2008 May 28.
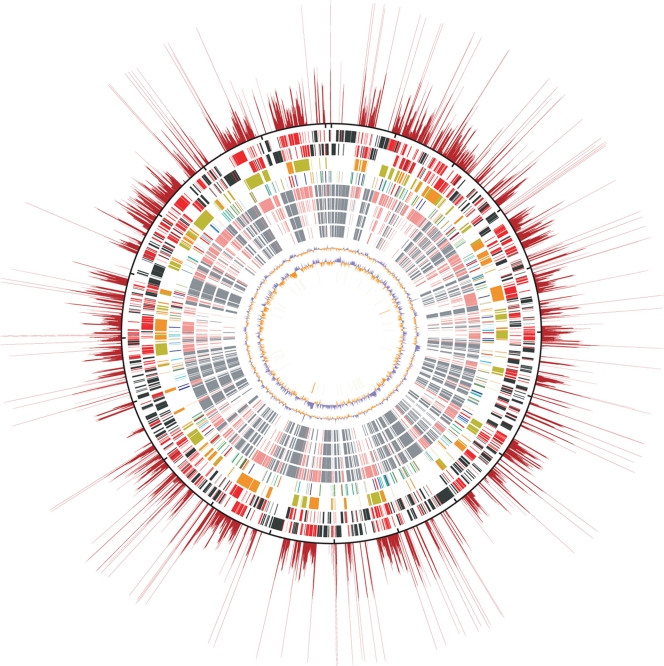
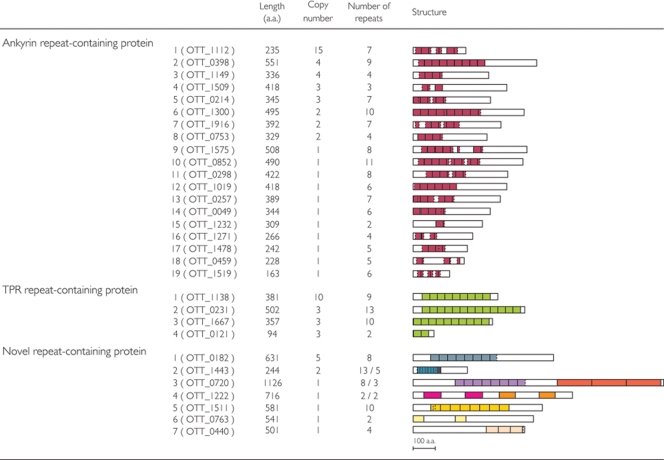
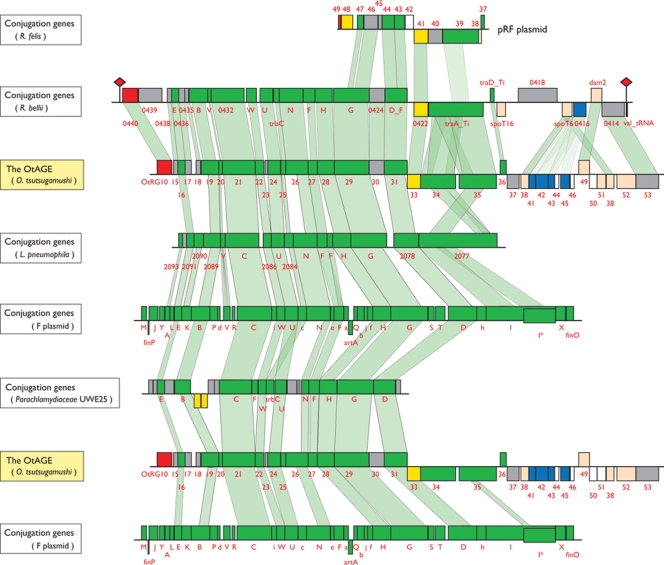
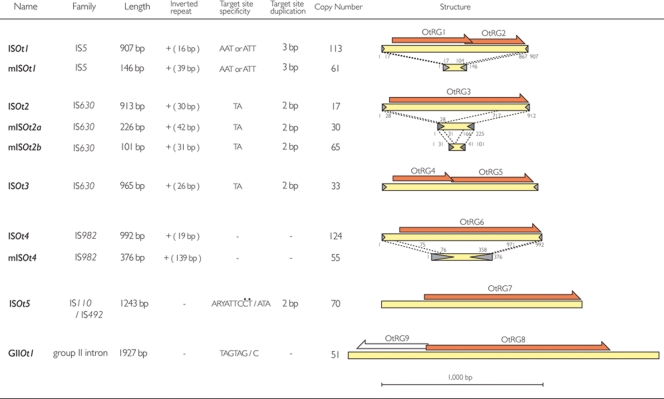
Scrub typhus ('Tsutsugamushi' disease in Japanese) is a mite-borne infectious disease. The causative agent is Orientia tsutsugamushi, an obligate intracellular bacterium belonging to the family Rickettsiaceae of the subdivision alpha-Proteobacteria. In this study, we determined the complete genome sequence of O. tsutsugamushi strain Ikeda, which comprises a single chromosome of 2 008 987 bp and contains 1967 protein coding sequences (CDSs). The chromosome is much larger than those of other members of Rickettsiaceae, and 46.7% of the sequence was occupied by repetitive sequences derived from an integrative and conjugative element, 10 types of transposable elements, and seven types of short repeats of unknown origins. The massive amplification and degradation of these elements have generated a huge number of repeated genes (1196 CDSs, categorized into 85 families), many of which are pseudogenes (766 CDSs), and also induced intensive genome shuffling. By comparing the gene content with those of other family members of Rickettsiacea, we identified the core gene set of the family Rickettsiaceae and found that, while much more extensive gene loss has taken place among the housekeeping genes of Orientia than those of Rickettsia, O. tsutsugamushi has acquired a large number of foreign genes. The O. tsutsugamushi genome sequence is thus a prominent example of the high plasticity of bacterial genomes, and provides the genetic basis for a better understanding of the biology of O. tsutsugamushi and the pathogenesis of 'Tsutsugamushi' disease.
恙虫病(日语中称为“Tsutsugamushi病”)是一种由螨传播的传染病。病原体是恙虫病东方体,它是一种专性细胞内细菌,属于α-变形菌门下的立克次氏体科。在本研究中,我们测定了恙虫病东方体池田株的完整基因组序列,该序列由一条2 008 987 bp的单一染色体组成,包含1967个蛋白质编码序列(CDS)。该染色体比立克次氏体科的其他成员的染色体大得多,46.7%的序列被来自整合和接合元件、10种转座元件以及7种未知来源的短重复序列的重复序列占据。这些元件的大量扩增和降解产生了大量重复基因(1196个CDS,分为85个家族),其中许多是假基因(766个CDS),还诱导了强烈的基因组重排。通过将基因内容与立克次氏体科其他家族成员的基因内容进行比较,我们确定了立克次氏体科的核心基因集,发现虽然东方体的管家基因比立克次氏体的管家基因发生了更广泛的基因丢失,但恙虫病东方体获得了大量外源基因。因此,恙虫病东方体的基因组序列是细菌基因组高度可塑性的一个突出例子,并为更好地理解恙虫病东方体的生物学特性和“Tsutsugamushi病”的发病机制提供了遗传基础。