Fregonese Laura, Stolk Jan
Alpha1 International Registry, c/o Department of Pulmology, Leiden University Medical Center, Leiden, The Netherlands.
Orphanet J Rare Dis. 2008 Jun 19;3:16. doi: 10.1186/1750-1172-3-16.
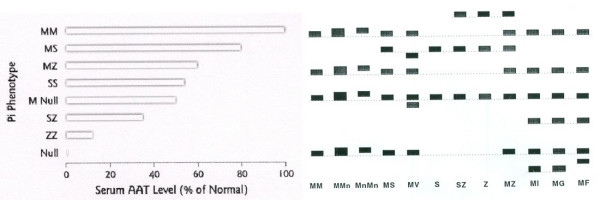
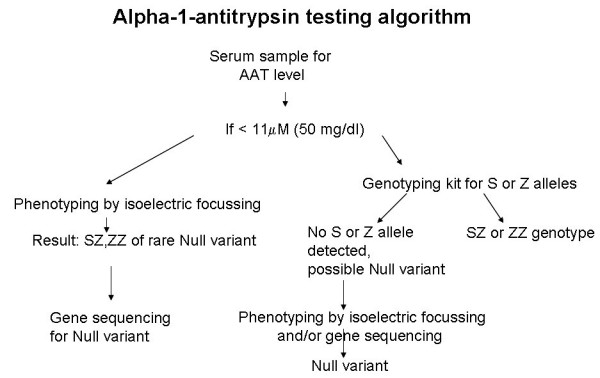
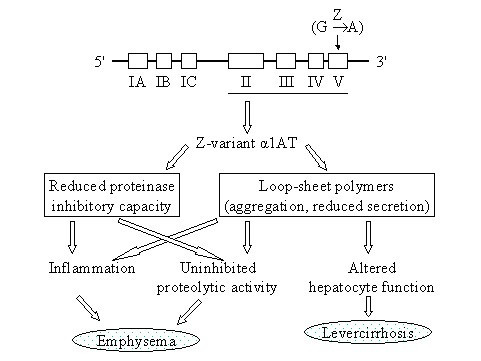
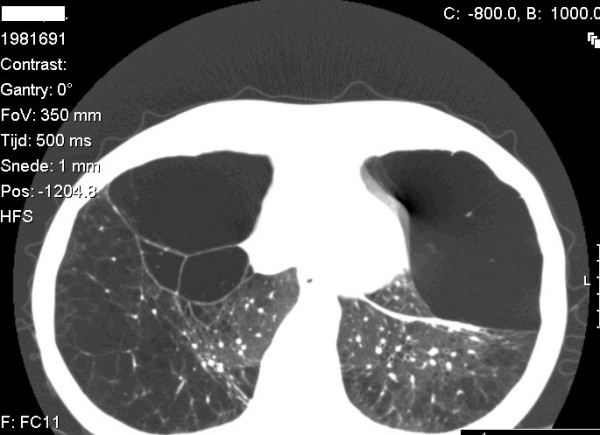
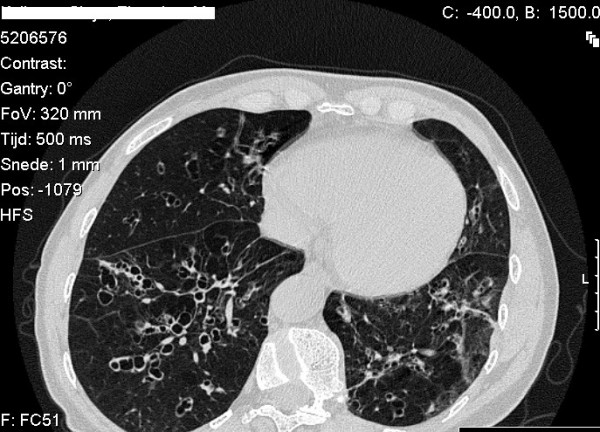
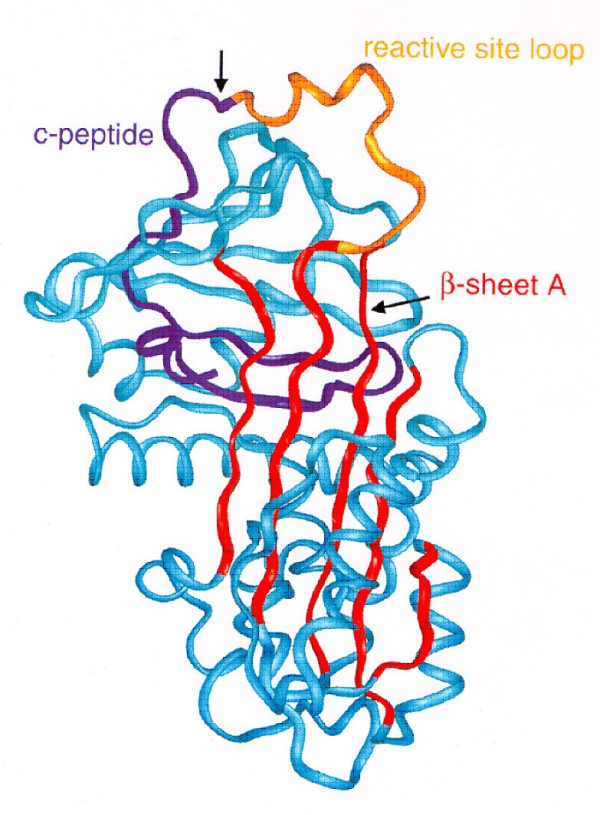
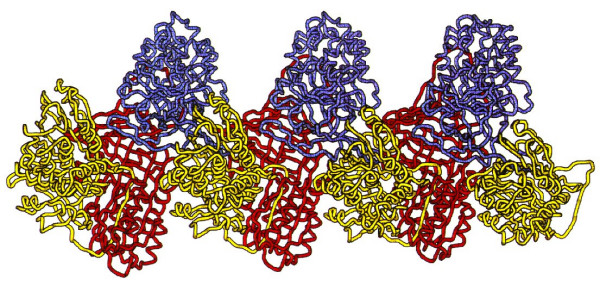
Alpha-1-antitrypsin deficiency (AATD) is a genetic disorder that manifests as pulmonary emphysema, liver cirrhosis and, rarely, as the skin disease panniculitis, and is characterized by low serum levels of AAT, the main protease inhibitor (PI) in human serum. The prevalence in Western Europe and in the USA is estimated at approximately 1 in 2,500 and 1 : 5,000 newborns, and is highly dependent on the Scandinavian descent within the population. The most common deficiency alleles in North Europe are PI Z and PI S, and the majority of individuals with severe AATD are PI type ZZ. The clinical manifestations may widely vary between patients, ranging from asymptomatic in some to fatal liver or lung disease in others. Type ZZ and SZ AATD are risk factors for the development of respiratory symptoms (dyspnoea, coughing), early onset emphysema, and airflow obstruction early in adult life. Environmental factors such as cigarette smoking, and dust exposure are additional risk factors and have been linked to an accelerated progression of this condition. Type ZZ AATD may also lead to the development of acute or chronic liver disease in childhood or adulthood: prolonged jaundice after birth with conjugated hyperbilirubinemia and abnormal liver enzymes are characteristic clinical signs. Cirrhotic liver failure may occur around age 50. In very rare cases, necrotizing panniculitis and secondary vasculitis may occur. AATD is caused by mutations in the SERPINA1 gene encoding AAT, and is inherited as an autosomal recessive trait. The diagnosis can be established by detection of low serum levels of AAT and isoelectric focusing. Differential diagnoses should exclude bleeding disorders or jaundice, viral infection, hemochromatosis, Wilson's disease and autoimmune hepatitis. For treatment of lung disease, intravenous alpha-1-antitrypsin augmentation therapy, annual flu vaccination and a pneumococcal vaccine every 5 years are recommended. Relief of breathlessness may be obtained with long-acting bronchodilators and inhaled corticosteroids. The end-stage liver and lung disease can be treated by organ transplantation. In AATD patients with cirrhosis, prognosis is generally grave.
α-1抗胰蛋白酶缺乏症(AATD)是一种遗传性疾病,表现为肺气肿、肝硬化,极少数情况下表现为皮肤疾病脂膜炎,其特征是血清中人类血清主要蛋白酶抑制剂(PI)α-1抗胰蛋白酶(AAT)水平较低。据估计,在西欧和美国,新生儿中的患病率约为2500分之一和5000分之一,并且高度依赖于人群中的斯堪的纳维亚血统。北欧最常见的缺陷等位基因是PI Z和PI S,大多数严重AATD患者为PI ZZ型。患者的临床表现差异很大,从某些人无症状到另一些人出现致命的肝脏或肺部疾病。ZZ型和SZ型AATD是出现呼吸道症状(呼吸困难、咳嗽)、早发性肺气肿以及成年早期气流阻塞的危险因素。吸烟和接触粉尘等环境因素是额外的危险因素,并且与这种疾病的加速进展有关。ZZ型AATD在儿童期或成年期也可能导致急性或慢性肝病的发生:出生后持续性黄疸伴结合胆红素血症和肝酶异常是其典型临床体征。肝硬化性肝衰竭可能在50岁左右发生。在极少数情况下,可能会出现坏死性脂膜炎和继发性血管炎。AATD由编码AAT的SERPINA1基因突变引起,呈常染色体隐性遗传。通过检测血清中AAT低水平和等电聚焦可确诊。鉴别诊断应排除出血性疾病或黄疸、病毒感染、血色素沉着症、威尔逊氏病和自身免疫性肝炎。对于肺部疾病的治疗,建议采用静脉注射α-1抗胰蛋白酶补充疗法、每年接种流感疫苗以及每5年接种一次肺炎球菌疫苗。长效支气管扩张剂和吸入性糖皮质激素可缓解呼吸困难。终末期肝病和肺病可通过器官移植治疗。对于患有肝硬化的AATD患者,预后通常不佳。