Zhou Jun, Fonseca Maria I, Pisalyaput Karntipa, Tenner Andrea J
Department of Molecular Biology and Biochemistry, Institute for Brain Aging and Dementia, Center for Immunology, University of California, Irvine, California 92697-3900, USA.
J Neurochem. 2008 Sep;106(5):2080-92. doi: 10.1111/j.1471-4159.2008.05558.x. Epub 2008 Jul 9.
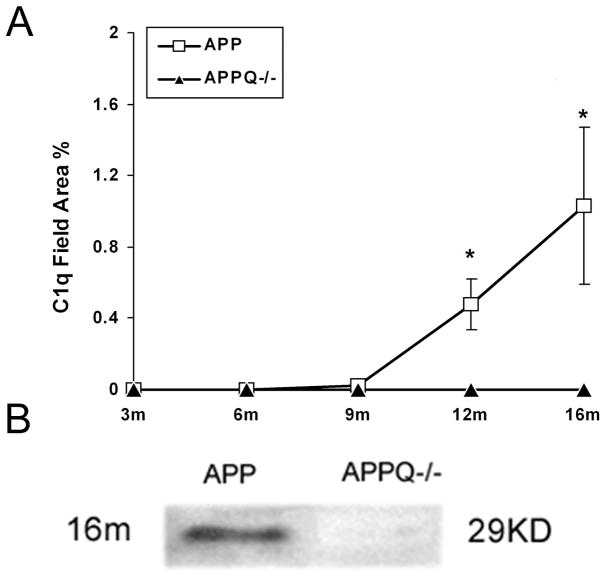
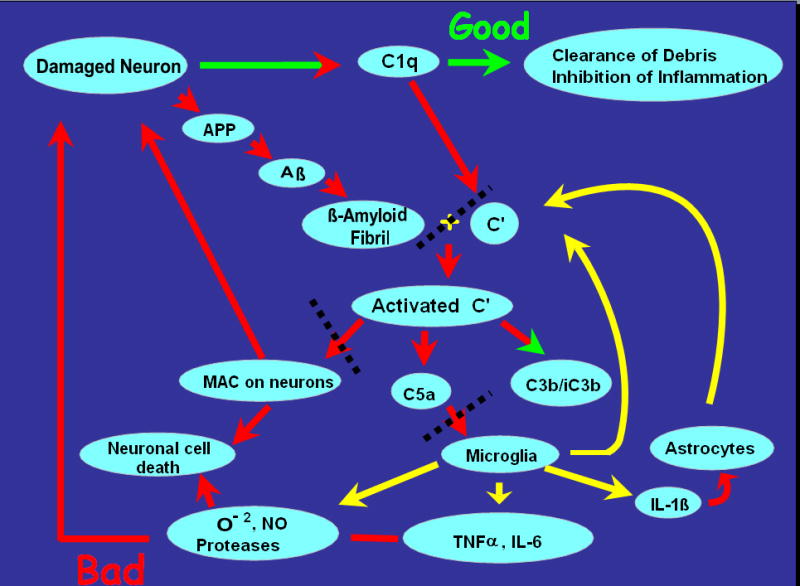
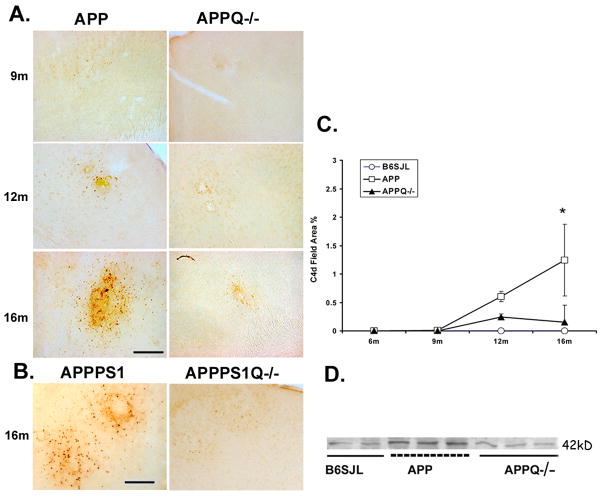
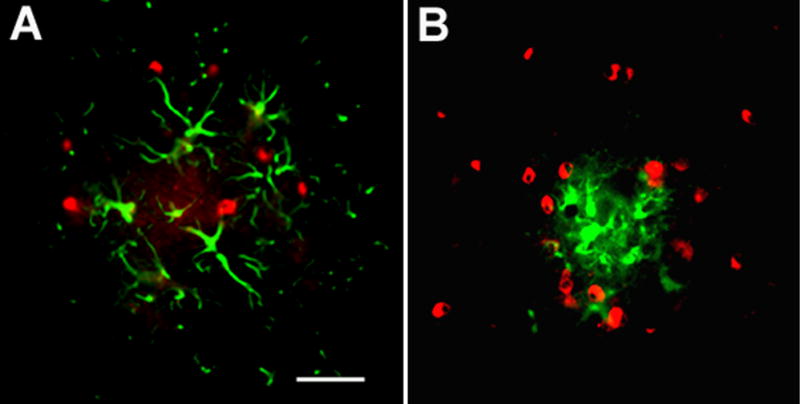
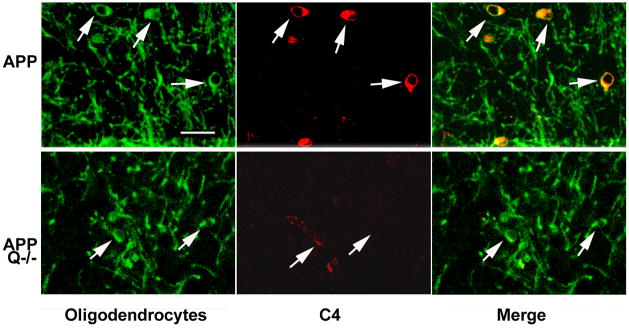
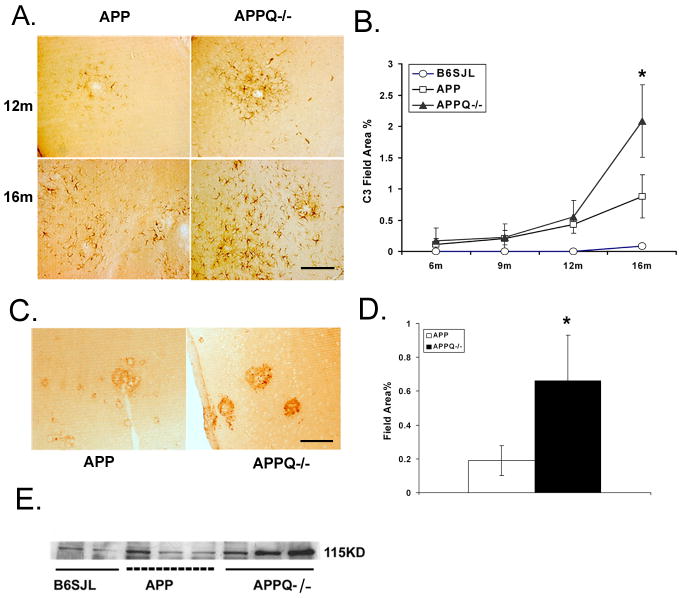
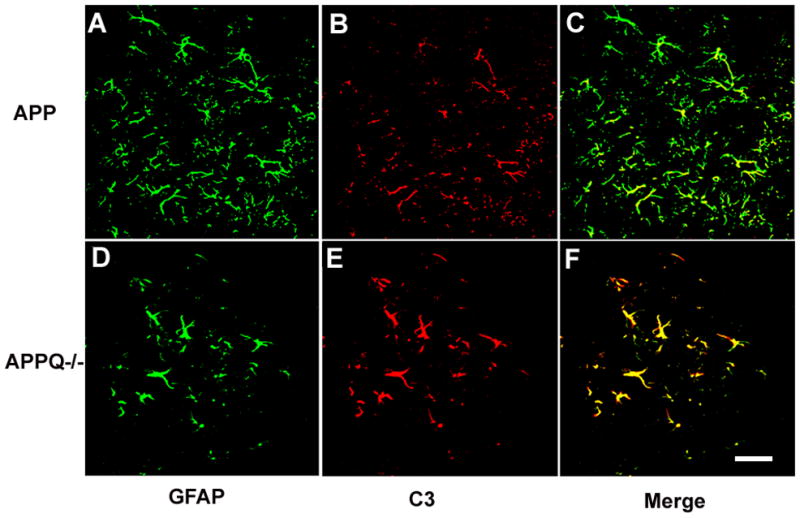
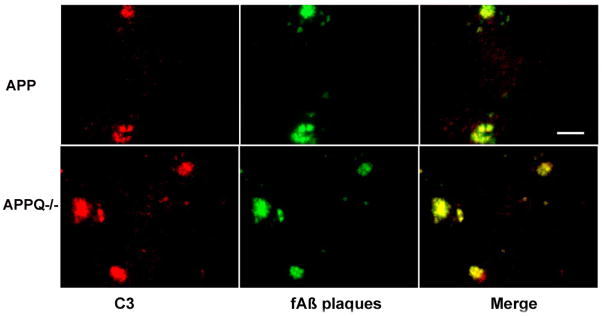
Alzheimer's disease (AD) is a neurodegenerative disease resulting in progressive cognitive decline. Amyloid plaque deposits consisting specifically of beta-amyloid peptides that have formed fibrils displaying beta-pleated sheet conformation are associated with activated microglia and astrocytes, are colocalized with C1q and other complement activation products, and appear at the time of cognitive decline in AD. Amyloid precursor protein (APP) transgenic mouse models of AD that lack the ability to activate the classical complement pathway display less neuropathology than do the APPQ+/+ mice, consistent with the hypothesis that complement activation and the resultant inflammation may play a role in the pathogenesis of AD. Further investigation of the presence of complement proteins C3 and C4 in the brain of these mice demonstrate that both C3 and C4 deposition increase with age in APPQ+/+ transgenic mice, as expected with the age-dependent increase in fibrillar beta-amyloid deposition. In addition, while C4 is predominantly localized on the plaques and/or associated with oligodendrocytes in APPQ+/+ mice, little C4 is detected in APPQ-/- brains consistent with a lack of classical complement pathway activation because of the absence of C1q in these mice. In contrast, plaque and cell associated C3 immunoreactivity is seen in both animal models and, surprisingly, is higher in APPQ-/- than in APPQ+/+ mice, providing evidence for alternative pathway activation. The unexpected increase in C3 levels in the APPQ-/- mice coincident with decreased neuropathology provides support for the hypothesis that complement can mediate protective events as well as detrimental events in this disease. Finally, induced expression of C3 in a subset of astrocytes suggests the existence of differential activation states of these cells.
阿尔茨海默病(AD)是一种导致进行性认知衰退的神经退行性疾病。由形成具有β-折叠片层构象的原纤维的β-淀粉样肽特异性组成的淀粉样斑块沉积物与活化的小胶质细胞和星形胶质细胞相关,与C1q和其他补体激活产物共定位,并在AD认知衰退时出现。缺乏激活经典补体途径能力的AD淀粉样前体蛋白(APP)转基因小鼠模型显示出比APPQ+/+小鼠更少的神经病理学特征,这与补体激活及由此产生的炎症可能在AD发病机制中起作用的假设一致。对这些小鼠大脑中补体蛋白C3和C4存在情况的进一步研究表明,在APPQ+/+转基因小鼠中,C3和C4沉积均随年龄增加,这与纤维状β-淀粉样蛋白沉积随年龄增长的情况相符。此外,虽然在APPQ+/+小鼠中C4主要定位于斑块和/或与少突胶质细胞相关,但在APPQ-/-大脑中几乎检测不到C4,这与这些小鼠因缺乏C1q而缺乏经典补体途径激活一致。相反,在两种动物模型中均可见斑块和细胞相关的C3免疫反应性,令人惊讶的是,APPQ-/-小鼠中的C3免疫反应性高于APPQ+/+小鼠,这为替代途径激活提供了证据。APPQ-/-小鼠中C3水平意外增加且神经病理学特征减少,这为补体在该疾病中既能介导有害事件又能介导保护事件的假设提供了支持。最后,在一部分星形胶质细胞中诱导C3表达表明这些细胞存在不同的激活状态。