Ng Pauline C, Levy Samuel, Huang Jiaqi, Stockwell Timothy B, Walenz Brian P, Li Kelvin, Axelrod Nelson, Busam Dana A, Strausberg Robert L, Venter J Craig
J Craig Venter Institute, Rockville, Maryland, United States of America.
PLoS Genet. 2008 Aug 15;4(8):e1000160. doi: 10.1371/journal.pgen.1000160.
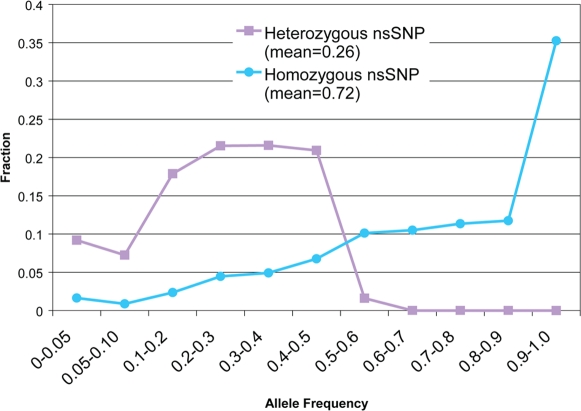
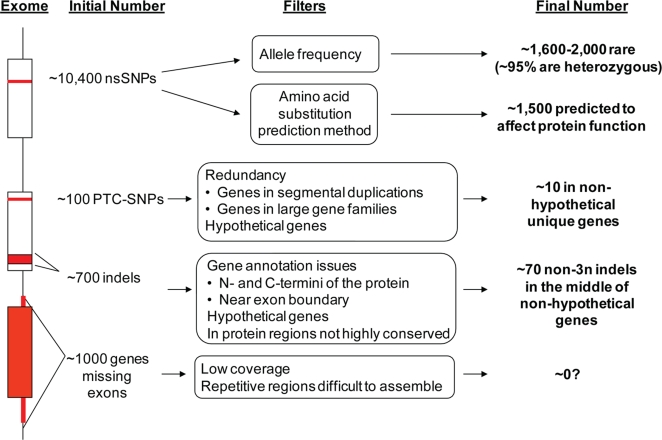
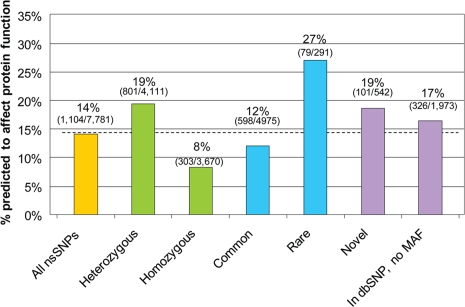
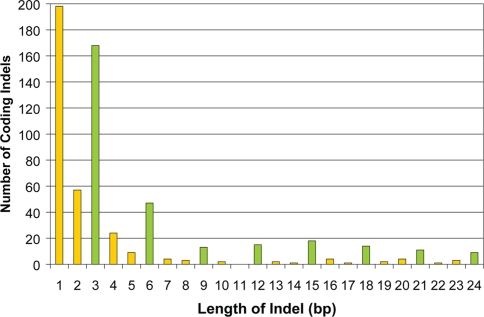
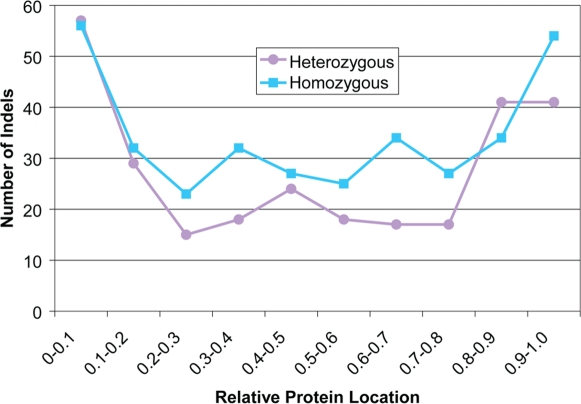
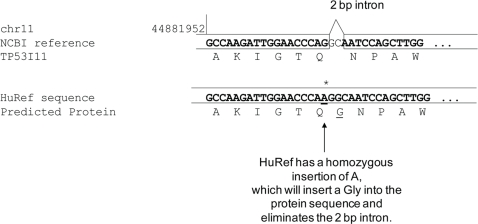
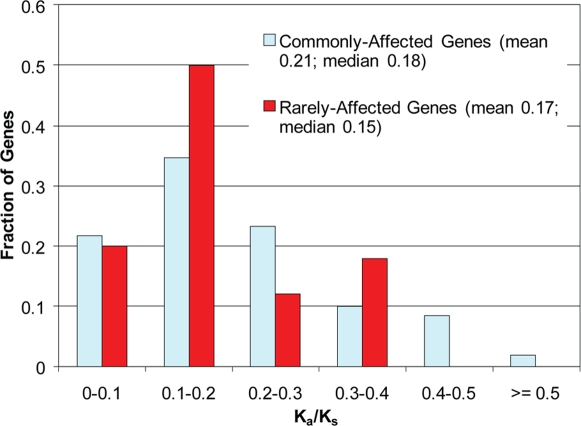
There is much interest in characterizing the variation in a human individual, because this may elucidate what contributes significantly to a person's phenotype, thereby enabling personalized genomics. We focus here on the variants in a person's 'exome,' which is the set of exons in a genome, because the exome is believed to harbor much of the functional variation. We provide an analysis of the approximately 12,500 variants that affect the protein coding portion of an individual's genome. We identified approximately 10,400 nonsynonymous single nucleotide polymorphisms (nsSNPs) in this individual, of which approximately 15-20% are rare in the human population. We predict approximately 1,500 nsSNPs affect protein function and these tend be heterozygous, rare, or novel. Of the approximately 700 coding indels, approximately half tend to have lengths that are a multiple of three, which causes insertions/deletions of amino acids in the corresponding protein, rather than introducing frameshifts. Coding indels also occur frequently at the termini of genes, so even if an indel causes a frameshift, an alternative start or stop site in the gene can still be used to make a functional protein. In summary, we reduced the set of approximately 12,500 nonsilent coding variants by approximately 8-fold to a set of variants that are most likely to have major effects on their proteins' functions. This is our first glimpse of an individual's exome and a snapshot of the current state of personalized genomics. The majority of coding variants in this individual are common and appear to be functionally neutral. Our results also indicate that some variants can be used to improve the current NCBI human reference genome. As more genomes are sequenced, many rare variants and non-SNP variants will be discovered. We present an approach to analyze the coding variation in humans by proposing multiple bioinformatic methods to hone in on possible functional variation.
人们对描述人类个体的变异非常感兴趣,因为这可能有助于阐明对一个人的表型有重大影响的因素,从而实现个性化基因组学。我们在此关注一个人的“外显子组”中的变异,外显子组是基因组中的外显子集合,因为人们认为外显子组包含了大部分功能变异。我们对大约12500个影响个体基因组蛋白质编码部分的变异进行了分析。我们在这个个体中鉴定出了大约10400个非同义单核苷酸多态性(nsSNP),其中大约15% - 20%在人类群体中是罕见的。我们预测大约1500个nsSNP会影响蛋白质功能,这些变异往往是杂合的、罕见的或新出现的。在大约700个编码插入缺失中,大约一半的长度往往是3的倍数,这会导致相应蛋白质中氨基酸的插入/缺失,而不是引入移码突变。编码插入缺失也经常出现在基因的末端,所以即使一个插入缺失导致了移码突变,基因中的一个替代起始或终止位点仍可用于产生功能性蛋白质。总之,我们将大约12500个非沉默编码变异的集合减少了约8倍,得到了一组最有可能对其蛋白质功能产生重大影响的变异。这是我们对个体外显子组的首次观察,也是个性化基因组学当前状态的一个快照。这个个体中的大多数编码变异是常见的,似乎在功能上是中性的。我们的结果还表明,一些变异可用于改进当前的NCBI人类参考基因组。随着更多基因组被测序,将会发现许多罕见变异和非SNP变异。我们通过提出多种生物信息学方法来专注于可能的功能变异,从而展示了一种分析人类编码变异的方法。