de Smith Adam J, Walters Robin G, Coin Lachlan J M, Steinfeld Israel, Yakhini Zohar, Sladek Rob, Froguel Philippe, Blakemore Alexandra I F
Section of Genomic Medicine, Imperial College London, Hammersmith Hospital, London, United Kingdom.
PLoS One. 2008 Aug 29;3(8):e3104. doi: 10.1371/journal.pone.0003104.
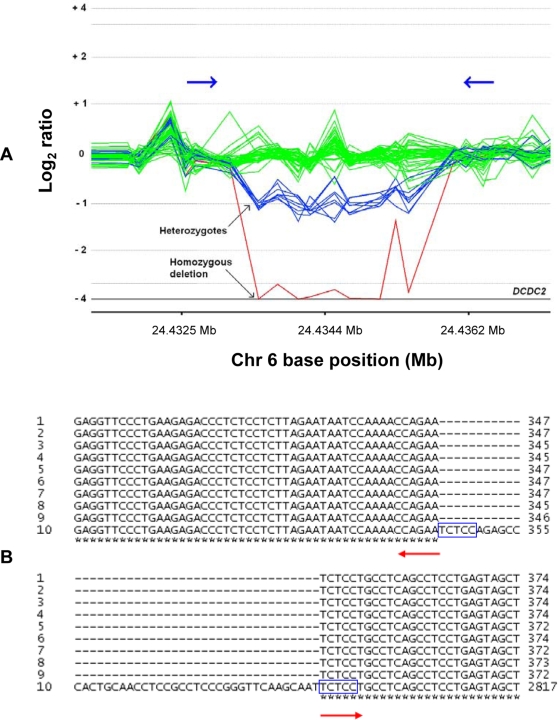
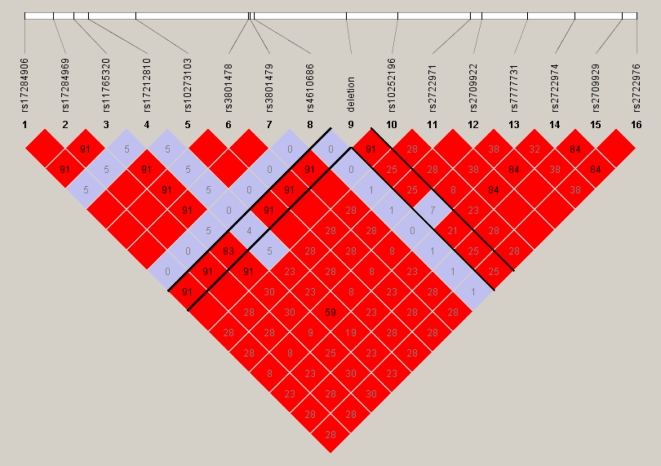
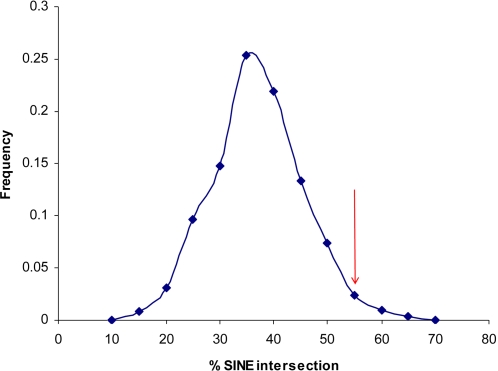
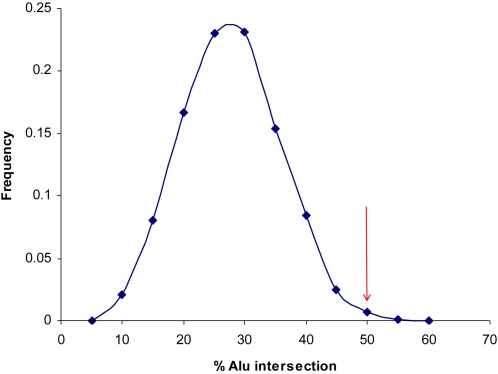
Copy number variants (CNVs) contribute significantly to human genomic variation, with over 5000 loci reported, covering more than 18% of the euchromatic human genome. Little is known, however, about the origin and stability of variants of different size and complexity. We investigated the breakpoints of 20 small, common deletions, representing a subset of those originally identified by array CGH, using Agilent microarrays, in 50 healthy French Caucasian subjects. By sequencing PCR products amplified using primers designed to span the deleted regions, we determined the exact size and genomic position of the deletions in all affected samples. For each deletion studied, all individuals carrying the deletion share identical upstream and downstream breakpoints at the sequence level, suggesting that the deletion event occurred just once and later became common in the population. This is supported by linkage disequilibrium (LD) analysis, which has revealed that most of the deletions studied are in moderate to strong LD with surrounding SNPs, and have conserved long-range haplotypes. Analysis of the sequences flanking the deletion breakpoints revealed an enrichment of microhomology at the breakpoint junctions. More significantly, we found an enrichment of Alu repeat elements, the overwhelming majority of which intersected deletion breakpoints at their poly-A tails. We found no enrichment of LINE elements or segmental duplications, in contrast to other reports. Sequence analysis revealed enrichment of a conserved motif in the sequences surrounding the deletion breakpoints, although whether this motif has any mechanistic role in the formation of some deletions has yet to be determined. Considered together with existing information on more complex inherited variant regions, and reports of de novo variants associated with autism, these data support the presence of different subgroups of CNV in the genome which may have originated through different mechanisms.
拷贝数变异(CNV)对人类基因组变异有重大贡献,已报道超过5000个位点,覆盖超过18%的常染色质人类基因组。然而,对于不同大小和复杂性变异的起源和稳定性知之甚少。我们使用安捷伦微阵列研究了50名健康法国白种人受试者中20个小的常见缺失的断点,这些缺失代表了最初通过阵列比较基因组杂交(array CGH)鉴定的一部分。通过对使用设计跨越缺失区域的引物扩增的PCR产物进行测序,我们确定了所有受影响样本中缺失的确切大小和基因组位置。对于所研究的每个缺失,所有携带该缺失的个体在序列水平上共享相同的上游和下游断点,这表明缺失事件仅发生一次,随后在人群中变得常见。连锁不平衡(LD)分析支持了这一点,该分析表明所研究的大多数缺失与周围的单核苷酸多态性(SNP)处于中度至强LD状态,并具有保守的长程单倍型。对缺失断点侧翼序列的分析揭示了断点连接处微同源性的富集。更重要的是,我们发现了Alu重复元件的富集,其中绝大多数在其多聚A尾处与缺失断点相交。与其他报道相反,我们未发现长散在核元件(LINE)或节段性重复的富集。序列分析揭示了缺失断点周围序列中一个保守基序的富集,尽管该基序在某些缺失形成中是否具有任何机制作用尚待确定。结合关于更复杂遗传变异区域的现有信息以及与自闭症相关的新发变异的报道,这些数据支持基因组中存在不同的CNV亚组,它们可能通过不同机制起源。