Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA.
Baylor Genetics, Houston, Texas 77021, USA.
Genome Res. 2018 Aug;28(8):1228-1242. doi: 10.1101/gr.229401.117. Epub 2018 Jun 15.
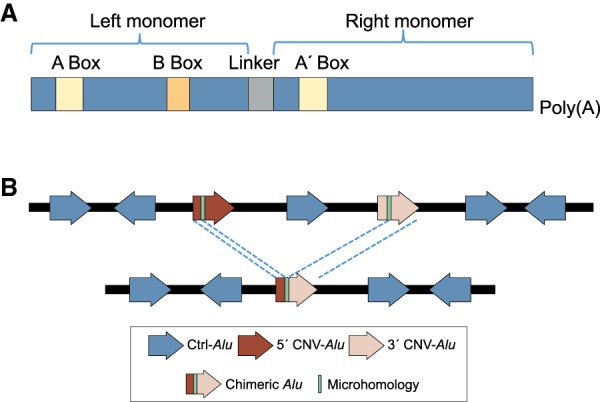
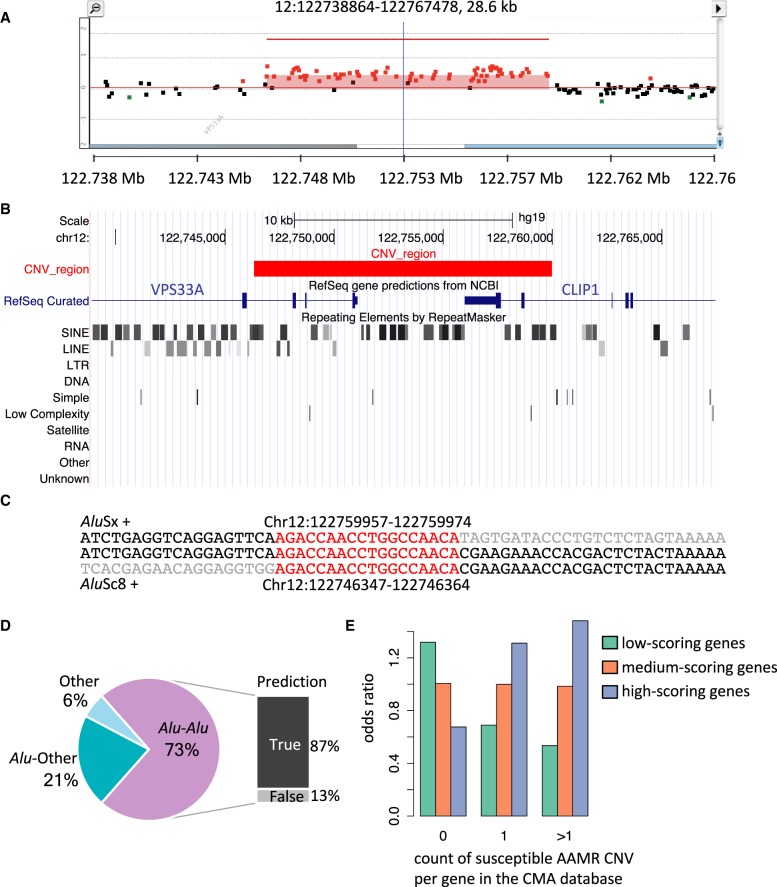
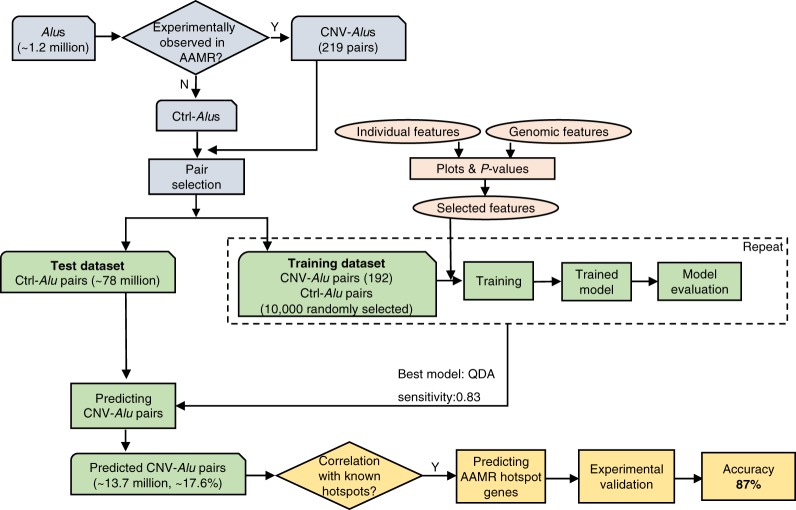
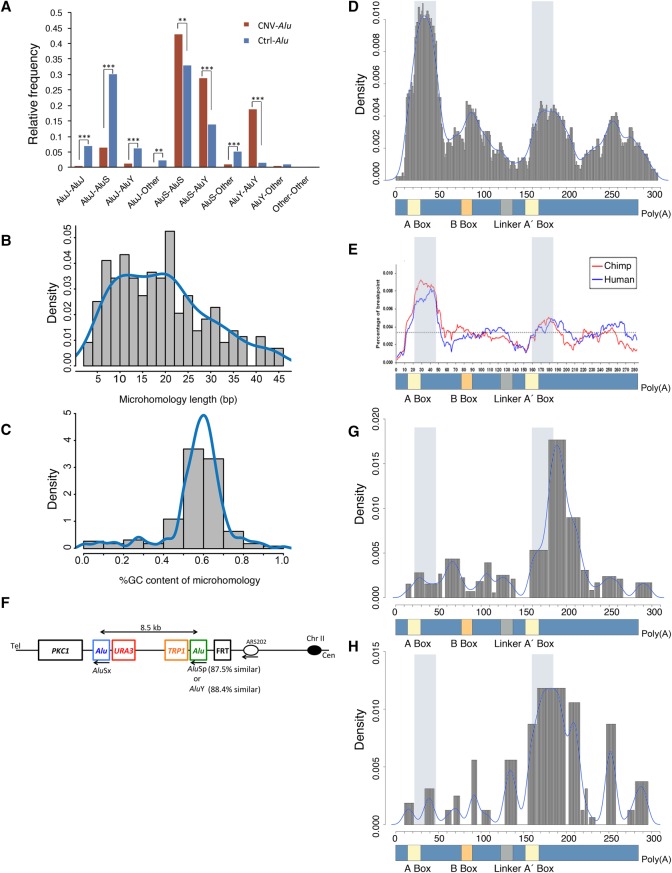
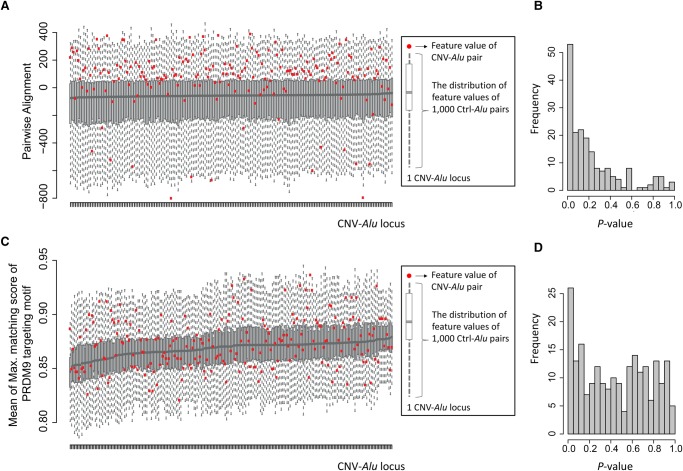
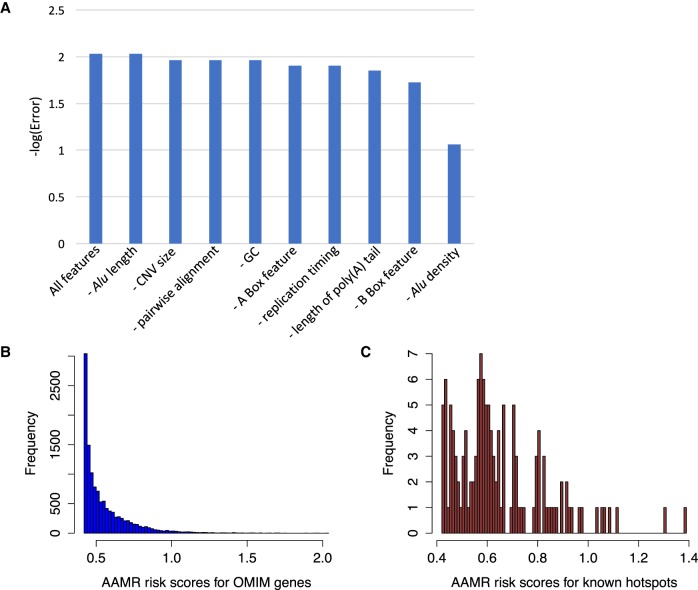
elements, the short interspersed element numbering more than 1 million copies per human genome, can mediate the formation of copy number variants (CNVs) between substrate pairs. These /-mediated rearrangements (AAMRs) can result in pathogenic variants that cause diseases. To investigate the impact of AAMR on gene variation and human health, we first characterized s that are involved in mediating CNVs (CNV-s) and observed that these s tend to be evolutionarily younger. We then computationally generated, with the assistance of a supercomputer, a test data set consisting of 78 million pairs and predicted ∼18% of them are potentially susceptible to AAMR. We further determined the relative risk of AAMR in 12,074 OMIM genes using the count of predicted CNV- pairs and experimentally validated the predictions with 89 samples selected by correlating predicted hotspots with a database of CNVs identified by clinical chromosomal microarrays (CMAs) on the genomes of approximately 54,000 subjects. We fine-mapped 47 duplications, 40 deletions, and two complex rearrangements and examined a total of 52 breakpoint junctions of simple CNVs. Overall, 94% of the candidate breakpoints were at least partially mediated. We successfully predicted all (100%) of pairs that mediated deletions ( = 21) and achieved an 87% positive predictive value overall when including AAMR-generated deletions and duplications. We provided a tool, AluAluCNVpredictor, for assessing AAMR hotspots and their role in human disease. These results demonstrate the utility of our predictive model and provide insights into the genomic features and molecular mechanisms underlying AAMR.
元件,短散布元件在人类基因组中数量超过 100 万份,可以介导底物对之间的拷贝数变异(CNVs)的形成。这些/介导的重排(AAMR)可导致导致疾病的致病性变异。为了研究 AAMR 对基因变异和人类健康的影响,我们首先对参与介导 CNVs(CNV-s)的 s 进行了特征描述,观察到这些 s 往往更年轻。然后,我们在超级计算机的协助下,通过计算生成了一个由 7800 万对组成的测试数据集,并预测其中约 18%可能易受 AAMR 影响。我们进一步通过预测的 CNV-对计数确定了 12074 个 OMIM 基因中的 AAMR 相对风险,并通过与临床染色体微阵列(CMA)确定的 CNV 数据库相关联,从 89 个样本中确定了预测热点,从而对预测结果进行了验证,这些样本是从大约 54000 个个体的基因组中选择的。我们对 47 个重复、40 个缺失和 2 个复杂重排进行了精细映射,并检查了 52 个简单 CNV 的总断点连接。总体而言,候选断点至少部分是/介导的。我们成功预测了所有(100%)介导缺失的对(=21),当包括 AAMR 生成的缺失和重复时,总体的阳性预测值达到 87%。我们提供了一种工具,AluAluCNVpredictor,用于评估 AAMR 热点及其在人类疾病中的作用。这些结果证明了我们预测模型的实用性,并提供了对 AAMR 背后的基因组特征和分子机制的深入了解。