Doniger Scott W, Kim Hyun Seok, Swain Devjanee, Corcuera Daniella, Williams Morgan, Yang Shiaw-Pyng, Fay Justin C
Computational Biology Program, Washington University, St. Louis, Missouri, United States of America.
PLoS Genet. 2008 Aug 29;4(8):e1000183. doi: 10.1371/journal.pgen.1000183.
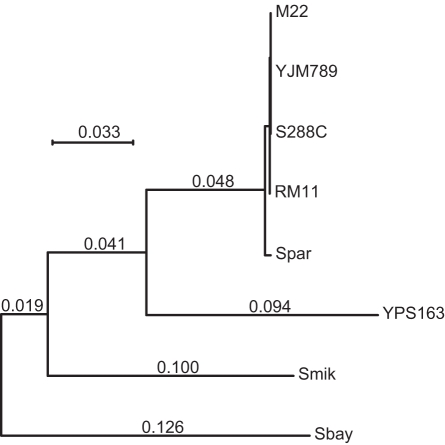
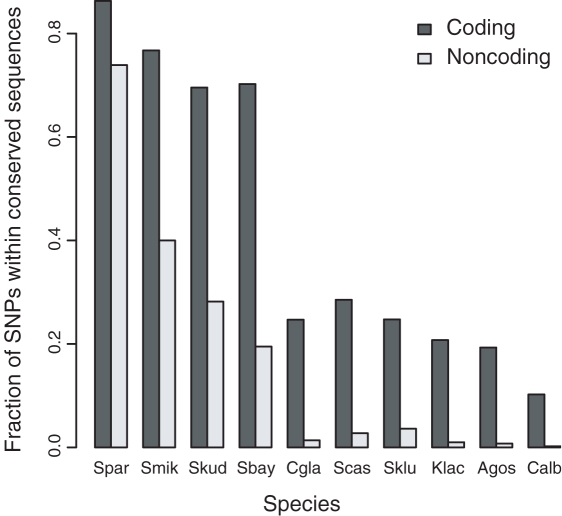
The abundance and identity of functional variation segregating in natural populations is paramount to dissecting the molecular basis of quantitative traits as well as human genetic diseases. Genome sequencing of multiple organisms of the same species provides an efficient means of cataloging rearrangements, insertion, or deletion polymorphisms (InDels) and single-nucleotide polymorphisms (SNPs). While inbreeding depression and heterosis imply that a substantial amount of polymorphism is deleterious, distinguishing deleterious from neutral polymorphism remains a significant challenge. To identify deleterious and neutral DNA sequence variation within Saccharomyces cerevisiae, we sequenced the genome of a vineyard and oak tree strain and compared them to a reference genome. Among these three strains, 6% of the genome is variable, mostly attributable to variation in genome content that results from large InDels. Out of the 88,000 polymorphisms identified, 93% are SNPs and a small but significant fraction can be attributed to recent interspecific introgression and ectopic gene conversion. In comparison to the reference genome, there is substantial evidence for functional variation in gene content and structure that results from large InDels, frame-shifts, and polymorphic start and stop codons. Comparison of polymorphism to divergence reveals scant evidence for positive selection but an abundance of evidence for deleterious SNPs. We estimate that 12% of coding and 7% of noncoding SNPs are deleterious. Based on divergence among 11 yeast species, we identified 1,666 nonsynonymous SNPs that disrupt conserved amino acids and 1,863 noncoding SNPs that disrupt conserved noncoding motifs. The deleterious coding SNPs include those known to affect quantitative traits, and a subset of the deleterious noncoding SNPs occurs in the promoters of genes that show allele-specific expression, implying that some cis-regulatory SNPs are deleterious. Our results show that the genome sequences of both closely and distantly related species provide a means of identifying deleterious polymorphisms that disrupt functionally conserved coding and noncoding sequences.
自然种群中分离的功能变异的丰度和特性对于剖析数量性状以及人类遗传疾病的分子基础至关重要。对同一物种的多个生物体进行基因组测序提供了一种有效的方法来编目重排、插入或缺失多态性(InDels)以及单核苷酸多态性(SNPs)。虽然近亲繁殖衰退和杂种优势意味着大量的多态性是有害的,但区分有害多态性和中性多态性仍然是一项重大挑战。为了鉴定酿酒酵母内的有害和中性DNA序列变异,我们对一株葡萄园菌株和一株橡树菌株的基因组进行了测序,并将它们与一个参考基因组进行比较。在这三个菌株中,6%的基因组是可变的,主要归因于由大InDels导致的基因组含量变异。在鉴定出的88,000个多态性中,93%是SNPs,一小部分但显著比例可归因于近期的种间渗入和异位基因转换。与参考基因组相比,有大量证据表明基因含量和结构的功能变异是由大InDels、移码以及多态性起始和终止密码子导致的。多态性与分歧的比较揭示了正向选择的证据很少,但有害SNPs的证据很多。我们估计12%的编码SNPs和7%的非编码SNPs是有害的。基于11个酵母物种之间的分歧,我们鉴定出1666个破坏保守氨基酸的非同义SNPs和1863个破坏保守非编码基序的非编码SNPs。有害的编码SNPs包括那些已知影响数量性状的,并且一部分有害的非编码SNPs出现在显示等位基因特异性表达的基因的启动子中,这意味着一些顺式调控SNPs是有害的。我们的结果表明,亲缘关系近和远的物种的基因组序列都提供了一种鉴定破坏功能保守的编码和非编码序列的有害多态性的方法。