Kulkarni Vinayak, Errami Mounir, Barber Robert, Garner Harold R
Mc Dermott Center for Human Growth and Development, UT Southwestern Medical Center, Dallas, TX, USA.
BMC Bioinformatics. 2008 Aug 12;9 Suppl 9(Suppl 9):S3. doi: 10.1186/1471-2105-9-S9-S3.
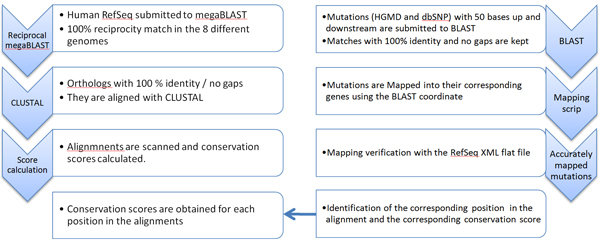
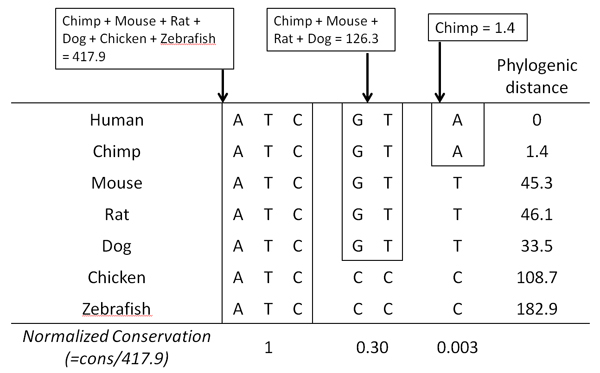
Single Nucleotide Polymorphisms (SNPs) are the most abundant form of genomic variation and can cause phenotypic differences between individuals, including diseases. Bases are subject to various levels of selection pressure, reflected in their inter-species conservation.
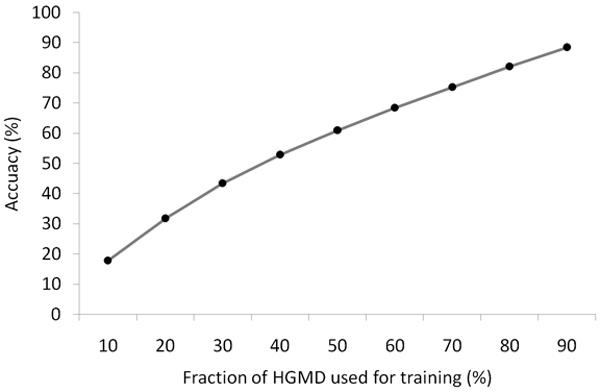
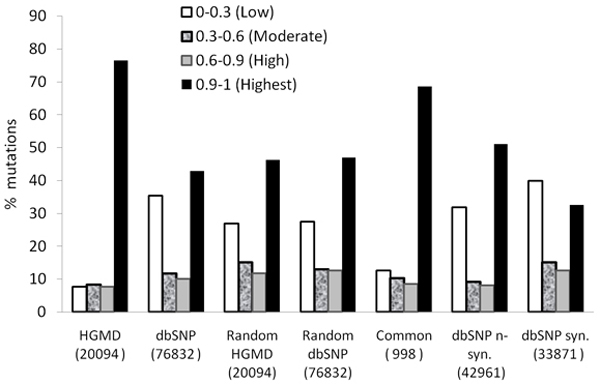
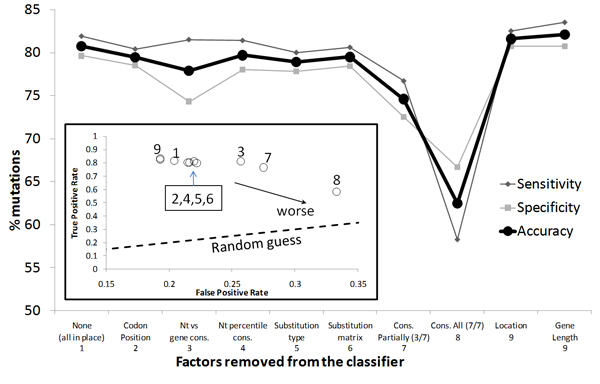
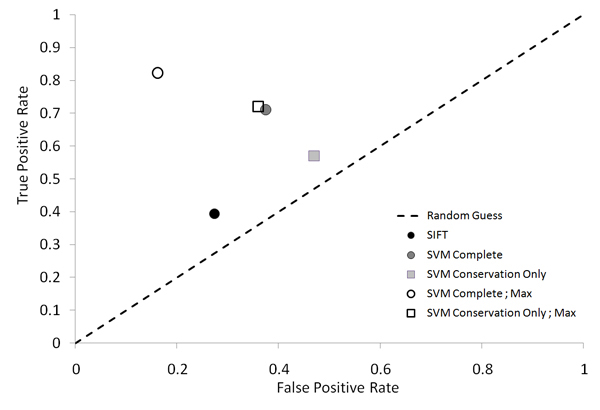
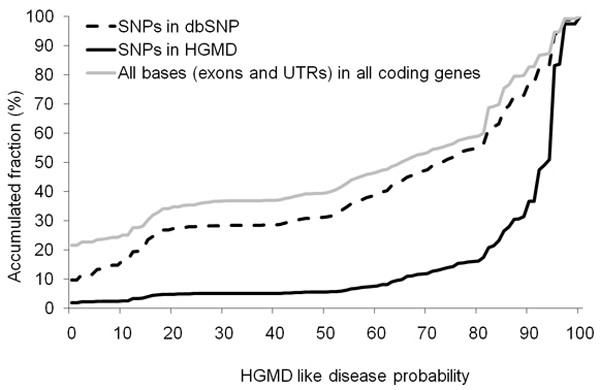
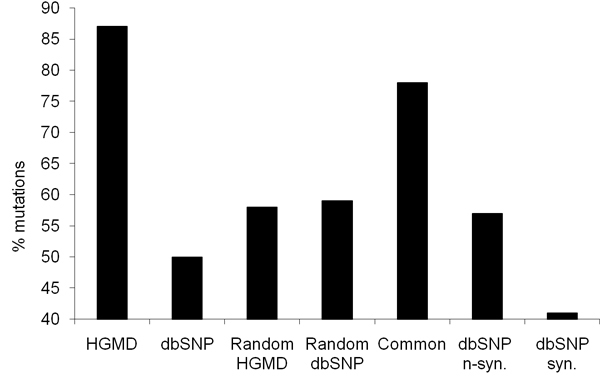
We propose a method that is not dependant on transcription information to score each coding base in the human genome reflecting the disease probability associated with its mutation. Twelve factors likely to be associated with disease alleles were chosen as the input for a support vector machine prediction algorithm. The analysis yielded 83% sensitivity and 84% specificity in segregating disease like alleles as found in the Human Gene Mutation Database from non-disease like alleles as found in the Database of Single Nucleotide Polymorphisms. This algorithm was subsequently applied to each base within all known human genes, exhaustively confirming that interspecies conservation is the strongest factor for disease association. For each gene, the length normalized average disease potential score was calculated. Out of the 30 genes with the highest scores, 21 are directly associated with a disease. In contrast, out of the 30 genes with the lowest scores, only one is associated with a disease as found in published literature. The results strongly suggest that the highest scoring genes are enriched for those that might contribute to disease, if mutated.
This method provides valuable information to researchers to identify sensitive positions in genes that have a high disease probability, enabling them to optimize experimental designs and interpret data emerging from genetic and epidemiological studies.
单核苷酸多态性(SNPs)是基因组变异最丰富的形式,可导致个体间的表型差异,包括疾病。碱基受到不同程度的选择压力,这在它们的种间保守性中得以体现。
我们提出了一种不依赖转录信息的方法,对人类基因组中的每个编码碱基进行评分,以反映与其突变相关的疾病概率。选择了十二个可能与疾病等位基因相关的因素作为支持向量机预测算法的输入。在将人类基因突变数据库中发现的疾病样等位基因与单核苷酸多态性数据库中发现的非疾病样等位基因区分开来时,该分析的灵敏度为83%,特异性为84%。随后将该算法应用于所有已知人类基因中的每个碱基,详尽地证实了种间保守性是疾病关联的最强因素。对于每个基因,计算了长度标准化的平均疾病潜力得分。得分最高的30个基因中,有21个与疾病直接相关。相比之下,得分最低的30个基因中,只有一个在已发表的文献中与疾病相关。结果强烈表明,得分最高的基因富集了那些如果发生突变可能导致疾病的基因。
该方法为研究人员提供了有价值的信息,以识别具有高疾病概率的基因中的敏感位置,使他们能够优化实验设计并解释遗传和流行病学研究中出现的数据。