Uchiyama Ikuo
Department of Theoretical Biology, National Institute for Basic Biology, National Institutes of Natural Sciences, Okazaki, Aichi, Japan.
BMC Genomics. 2008 Oct 31;9:515. doi: 10.1186/1471-2164-9-515.
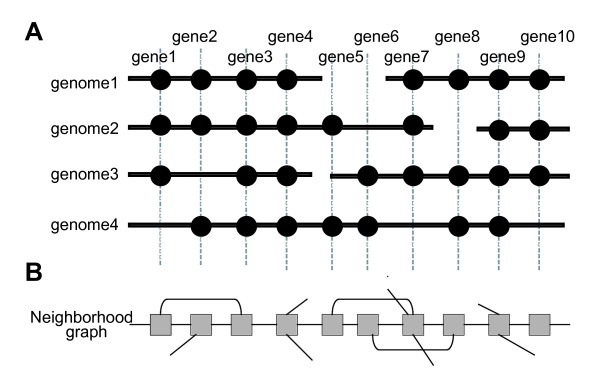
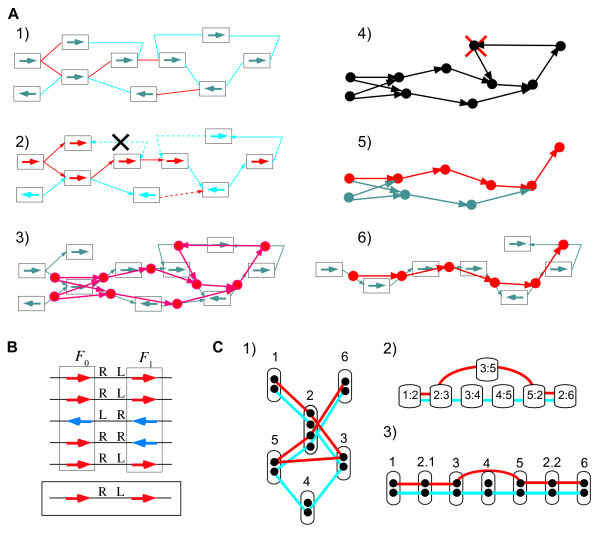
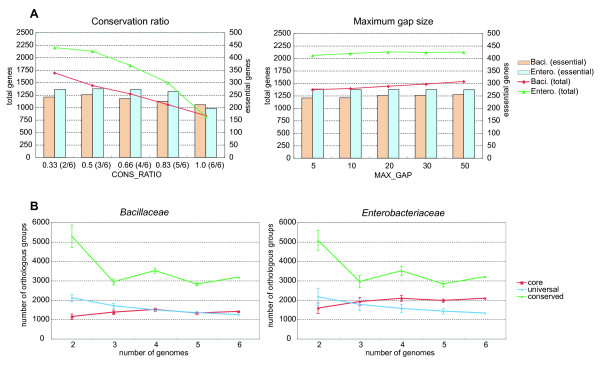
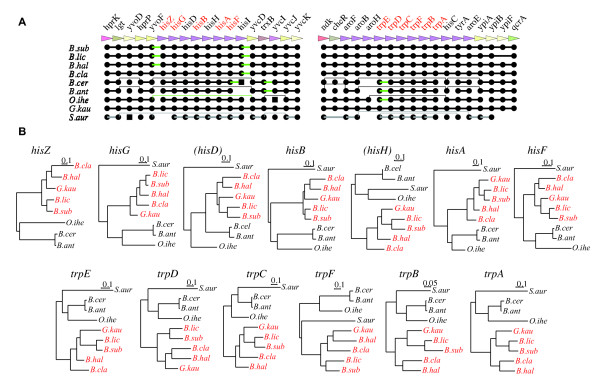
Identifying the set of intrinsically conserved genes, or the genomic core, among related genomes is crucial for understanding prokaryotic genomes where horizontal gene transfers are common. Although core genome identification appears to be obvious among very closely related genomes, it becomes more difficult when more distantly related genomes are compared. Here, we consider the core structure as a set of sufficiently long segments in which gene orders are conserved so that they are likely to have been inherited mainly through vertical transfer, and developed a method for identifying the core structure by finding the order of pre-identified orthologous groups (OGs) that maximally retains the conserved gene orders.
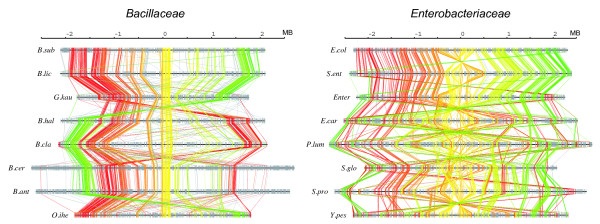
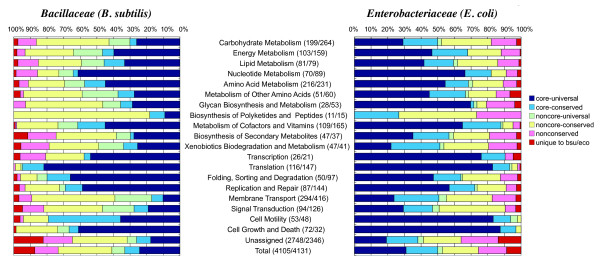
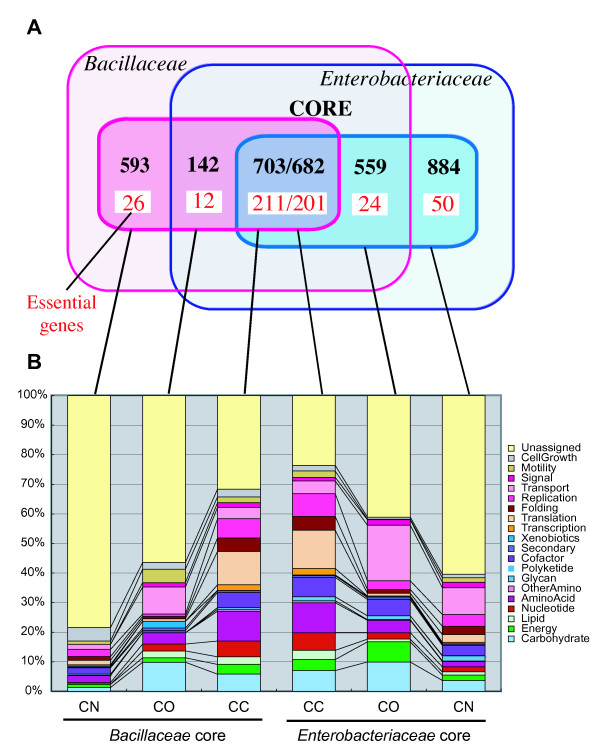
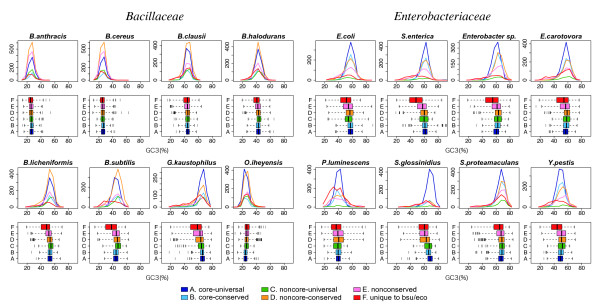
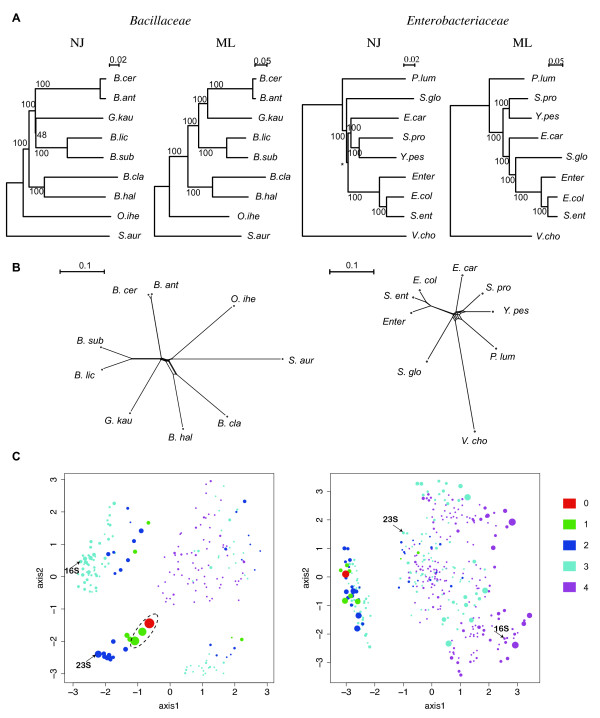
The method was applied to genome comparisons of two well-characterized families, Bacillaceae and Enterobacteriaceae, and identified their core structures comprising 1438 and 2125 OGs, respectively. The core sets contained most of the essential genes and their related genes, which were primarily included in the intersection of the two core sets comprising around 700 OGs. The definition of the genomic core based on gene order conservation was demonstrated to be more robust than the simpler approach based only on gene conservation. We also investigated the core structures in terms of G+C content homogeneity and phylogenetic congruence, and found that the core genes primarily exhibited the expected characteristic, i.e., being indigenous and sharing the same history, more than the non-core genes.
The results demonstrate that our strategy of genome alignment based on gene order conservation can provide an effective approach to identify the genomic core among moderately related microbial genomes.
在相关基因组中识别内在保守基因集或基因组核心,对于理解水平基因转移常见的原核生物基因组至关重要。尽管在亲缘关系非常近的基因组中,核心基因组的识别似乎很明显,但在比较亲缘关系较远的基因组时,难度会增加。在这里,我们将核心结构视为一组足够长的片段,其中基因顺序是保守的,因此它们很可能主要通过垂直转移遗传而来,并开发了一种方法,通过找到预先识别的直系同源组(OGs)的顺序来识别核心结构,该顺序能最大程度地保留保守的基因顺序。
该方法应用于两个特征明确的科(芽孢杆菌科和肠杆菌科)的基因组比较,分别识别出它们的核心结构包含1438个和2125个OGs。核心集包含了大部分必需基因及其相关基因,这些基因主要包含在两个核心集的交集中,约有700个OGs。基于基因顺序保守性的基因组核心定义被证明比仅基于基因保守性的更简单方法更稳健。我们还从G+C含量同质性和系统发育一致性方面研究了核心结构,发现核心基因比非核心基因更主要地表现出预期特征,即具有本土性且共享相同历史。
结果表明,我们基于基因顺序保守性的基因组比对策略能够为识别中等亲缘关系的微生物基因组中的基因组核心提供一种有效方法。